当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
单层Cu2Se上的原子吸附:第一性原理研究
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2021-3-26 , DOI: 10.1039/d1cp00169h
Yizhou You 1, 2, 3, 4, 5 , Huimin Hu 1, 2, 3, 4, 5 , Jin-Ho Choi 1, 2, 3, 4, 5
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2021-3-26 , DOI: 10.1039/d1cp00169h
Yizhou You 1, 2, 3, 4, 5 , Huimin Hu 1, 2, 3, 4, 5 , Jin-Ho Choi 1, 2, 3, 4, 5
Affiliation
|
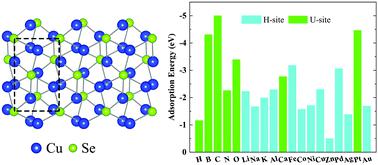
单层Cu 2 Se是一种新型的二维(2D)材料,但其基本性能尚未得到充分研究。因此,在这项工作中,我们使用第一原理密度泛函理论计算来研究各种元素在单层Cu 2 Se上的吸附行为。所考虑的元素包括金属(Li,Na,Al,K,Ca,Fe,Co,Ni,Cu,Zn,Pd,Ag,Pt和Au)和非金属(H,B,C,N和O)。所有这些原子的吸附都是放热的,具有大量的结合能。尽管单层Cu 2 Se与所有被吸附物形成强键,但仍保留其层状结构。这种原子吸附实质上改变了2D Cu 2的电子性质。硒 尤其是,N,Fe,Co,Ni和Au原子会在单层Cu 2 Se的带隙内产生中带隙态。此外,除了金以外,其他原子也表现出磁矩。自然地,这种电子结构改性还导致单层Cu 2 Se的功函数的改变。本工作表明,原子吸附可以优化单层Cu 2 Se的性能。

"点击查看英文标题和摘要"
更新日期:2021-04-14
"点击查看英文标题和摘要"
































 京公网安备 11010802027423号
京公网安备 11010802027423号