背景与目标
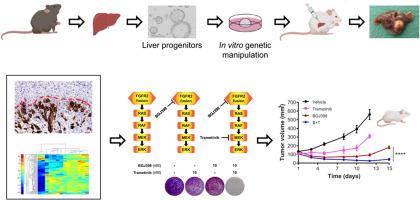
大约 15% 的肝内胆管癌 (iCCA) 表达成纤维细胞生长因子受体 2 (FGFR2) 融合蛋白 (FFs),通常伴随着TP53、CDKN2A或BAP1的突变失活。在 FF 中,FGFR2 残基 1-768 与由多种伴侣基因 (>60) 编码的序列融合,导致致癌 FF 激活。虽然 FGFR 特异性酪氨酸激酶抑制剂 (F-TKI) 在 FF +iCCA,反应是部分的和/或受抗性机制限制,例如 FGFR2 看门人残基中的 V565F 替代。因此,改进 iCCA 中的 FF 目标仍然是一个关键的未满足需求。在这里,我们旨在生成 FF 驱动的 iCCA 的小鼠模型,并使用它来发现可操作的 FF 相关依赖项。
方法
四个携带不同融合序列的 iCCA FF 在Tp53 -/-小鼠肝脏类器官中表达。通过移植到免疫缺陷小鼠中评估转基因肝脏类器官的致瘤特性。来自肿瘤性病变的细胞模型被用于临床前研究。
结果
根据组织学、表型和转录组学分析,移植表达 FF 的肝脏类器官产生了被诊断为 CCA 的肿瘤。这种致瘤表型的外显率受 FF 身份的影响。无论 FF 身份或 V565F 突变如何,来自 CCA 病变的肿瘤类器官和 2D 细胞系都沉迷于通过 Ras-Erk 进行的 FF 信号传导。通过同时对 FF 和 Mek1/2 进行药理抑制,对 FF 和 Ras-Erk 通路的双重阻断在体外和体内提供了比单药 F-TKI 更大的治疗效果。
结论
FF 驱动的 iCCA 发病机制成功地模拟了Tp53 -/-小鼠背景,揭示了结构不同的 FF 之间的生物异质性。FF-ERK 信号的双重阻断值得考虑针对人类 FF + iCCA 的基于精度的方法。
总结
肝内胆管癌 (iCCA) 是一种难以治疗的罕见癌症。iCCA 的一种亚型是由产生称为 FGFR2 融合的致癌驱动因子的基因组改变引起的。FGFR2 融合患者对 FGFR 抑制剂有反应,但临床反应通常持续时间适中。我们使用动物和细胞模型来证明 FGFR2 融合需要名为 Mek1/2 的下游效应器的活性。我们发现双重阻断 FGFR2 融合和 Mek1/2 比单独抑制 FGFR2 融合更有效,表明双重阻断 FGFR2-MEK1/2 在 iCCA 患者中的潜在临床应用。
 "点击查看英文标题和摘要"
"点击查看英文标题和摘要"
FGFR2 fusion proteins drive oncogenic transformation of mouse liver organoids towards cholangiocarcinoma
Background & Aims
About 15% of intrahepatic cholangiocarcinomas (iCCAs) express fibroblast growth factor receptor 2 (FGFR2) fusion proteins (FFs), usually alongside mutational inactivation of TP53, CDKN2A or BAP1. In FFs, FGFR2 residues 1-768 fuse to sequences encoded by a diverse array of partner genes (>60) causing oncogenic FF activation. While FGFR-specific tyrosine kinase inhibitors (F-TKI) provide clinical benefit in FF+ iCCA, responses are partial and/or limited by resistance mechanisms, such as the V565F substitution in the FGFR2 gatekeeper residue. Improving on FF targeting in iCCA therefore remains a critical unmet need. Herein, we aimed to generate a murine model of FF-driven iCCA and use this to uncover actionable FF-associated dependencies.
Methods
Four iCCA FFs carrying different fusion sequences were expressed in Tp53-/- mouse liver organoids. Tumorigenic properties of genetically modified liver organoids were assessed by transplantation into immuno-deficient mice. Cellular models derived from neoplastic lesions were exploited for pre-clinical studies.
Results
Transplantation of FF-expressing liver organoids yielded tumors diagnosed as CCA based on histological, phenotypic and transcriptomic analyses. The penetrance of this tumorigenic phenotype was influenced by FF identity. Tumor organoids and 2D cell lines derived from CCA lesions were addicted to FF signaling via Ras-Erk, regardless of FF identity or V565F mutation. Dual blockade of FF and the Ras-Erk pathway by concomitant pharmacological inhibition of FFs and Mek1/2 provided greater therapeutic efficacy than single agent F-TKI in vitro and in vivo.
Conclusions
FF-driven iCCA pathogenesis was successfully modeled on a Tp53-/- murine background, revealing biological heterogeneity among structurally different FFs. Double blockade of FF-ERK signaling deserves consideration for precision-based approaches against human FF+ iCCA.
Lay summary
Intrahepatic cholangiocarcinoma (iCCA) is a rare cancer that is difficult to treat. A subtype of iCCA is caused by genomic alterations that generate oncogenic drivers known as FGFR2 fusions. Patients with FGFR2 fusions respond to FGFR inhibitors, but clinical responses are often of modest duration. We used animal and cellular models to show that FGFR2 fusions require the activity of a downstream effector named Mek1/2. We found that dual blockade of FGFR2 fusions and Mek1/2 was more effective than isolated inhibition of FGFR2 fusions, pointing to the potential clinical utility of dual FGFR2-MEK1/2 blockade in patients with iCCA.
































 京公网安备 11010802027423号
京公网安备 11010802027423号