当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Developing a variation of 3D‐QSAR/MD method in drug design
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2021-03-14 , DOI: 10.1002/jcc.26514
Hamed Haghshenas 1 , Bita Kaviani 2 , Monireh Firouzeh 3 , Hossein Tavakol 4
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2021-03-14 , DOI: 10.1002/jcc.26514
Hamed Haghshenas 1 , Bita Kaviani 2 , Monireh Firouzeh 3 , Hossein Tavakol 4
Affiliation
|
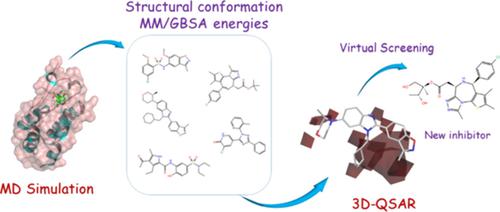
In continuation of the previous reports on a combination of 3D‐quantitative structure–activity relationships (QSAR) with computational molecular dynamics (MD) studies, a new variation of 3D‐QSAR/MD method has been employed for drug‐design as an alternative or supplementary for the typical experimental methods. The presented method is more cost‐effective and less time‐consuming than the previous methods and avoids several restrictions of experimental methods, such as validity estimation, and predictability. For this purpose, seven inhibitors for bromodomain (BRD)‐containing protein, as an important protein in the development of different types of cancer and responsible for oncogenic rearrangements, have been selected to study of their interactions by docking and MD simulations using molecular mechanics/generalized born surface area (MM/GBSA) method. To build the proposed model, a common variant of 3D‐QSAR methods, comparative molecular field analysis has been employed using a dataset of 100 MD‐extracted ligand conformations and their corresponding MM/GBSA BRD4‐binding energies. The results showed excellent predictability of the generated model for both the training set and test groups. Finally, two new inhibitors were selected among total 4000 designed derivatives (generated through evolutionary techniques) using the proposed 3D‐QSAR‐MD model. The potentials of these inhibitors were assessed by MD simulations, which showed the higher inhibitory of these compounds than the previous inhibitors. Therefore, this method showed high potentials for acceleration of the procedure of drug design and a basis for joining researchers in computational biology and pharmaceutical sciences.
中文翻译:

在药物设计中开发 3D-QSAR/MD 方法的变体
继先前关于 3D 定量结构-活性关系 (QSAR) 与计算分子动力学 (MD) 研究相结合的报告之后,3D-QSAR/MD 方法的新变体已被用于药物设计作为替代或对典型实验方法的补充。所提出的方法比以前的方法更具成本效益且耗时更少,并且避免了实验方法的一些限制,例如有效性估计和可预测性。为此,我们选择了七种含溴结构域 (BRD) 蛋白抑制剂,作为不同类型癌症发展中的重要蛋白质,并负责致癌重排,已被选择用于通过对接和使用分子力学的 MD 模拟来研究它们的相互作用。广义出生表面积(MM/GBSA)法。为了构建所提出的模型,这是 3D-QSAR 方法的一种常见变体,使用了 100 个 MD 提取的配体构象及其相应的 MM/GBSA BRD4 结合能的数据集进行了比较分子场分析。结果表明,生成的模型对于训练集和测试组都具有出色的可预测性。最后,使用所提出的 3D-QSAR-MD 模型从总共 4000 个设计的衍生物(通过进化技术生成)中选择了两种新的抑制剂。这些抑制剂的潜力通过 MD 模拟进行评估,表明这些化合物的抑制作用比以前的抑制剂更高。因此,该方法显示出加速药物设计过程的巨大潜力,并为加入计算生物学和药物科学领域的研究人员奠定了基础。
更新日期:2021-04-08
中文翻译:
在药物设计中开发 3D-QSAR/MD 方法的变体
继先前关于 3D 定量结构-活性关系 (QSAR) 与计算分子动力学 (MD) 研究相结合的报告之后,3D-QSAR/MD 方法的新变体已被用于药物设计作为替代或对典型实验方法的补充。所提出的方法比以前的方法更具成本效益且耗时更少,并且避免了实验方法的一些限制,例如有效性估计和可预测性。为此,我们选择了七种含溴结构域 (BRD) 蛋白抑制剂,作为不同类型癌症发展中的重要蛋白质,并负责致癌重排,已被选择用于通过对接和使用分子力学的 MD 模拟来研究它们的相互作用。广义出生表面积(MM/GBSA)法。为了构建所提出的模型,这是 3D-QSAR 方法的一种常见变体,使用了 100 个 MD 提取的配体构象及其相应的 MM/GBSA BRD4 结合能的数据集进行了比较分子场分析。结果表明,生成的模型对于训练集和测试组都具有出色的可预测性。最后,使用所提出的 3D-QSAR-MD 模型从总共 4000 个设计的衍生物(通过进化技术生成)中选择了两种新的抑制剂。这些抑制剂的潜力通过 MD 模拟进行评估,表明这些化合物的抑制作用比以前的抑制剂更高。因此,该方法显示出加速药物设计过程的巨大潜力,并为加入计算生物学和药物科学领域的研究人员奠定了基础。
































 京公网安备 11010802027423号
京公网安备 11010802027423号