Cell ( IF 45.5 ) Pub Date : 2021-02-18 , DOI: 10.1016/j.cell.2021.01.029 Luis F Camarillo-Guerrero 1 , Alexandre Almeida 2 , Guillermo Rangel-Pineros 3 , Robert D Finn 4 , Trevor D Lawley 1
|
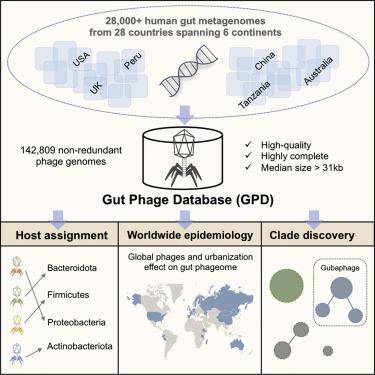
Bacteriophages drive evolutionary change in bacterial communities by creating gene flow networks that fuel ecological adaptions. However, the extent of viral diversity and its prevalence in the human gut remains largely unknown. Here, we introduce the Gut Phage Database, a collection of ∼142,000 non-redundant viral genomes (>10 kb) obtained by mining a dataset of 28,060 globally distributed human gut metagenomes and 2,898 reference genomes of cultured gut bacteria. Host assignment revealed that viral diversity is highest in the Firmicutes phyla and that ∼36% of viral clusters (VCs) are not restricted to a single species, creating gene flow networks across phylogenetically distinct bacterial species. Epidemiological analysis uncovered 280 globally distributed VCs found in at least 5 continents and a highly prevalent phage clade with features reminiscent of p-crAssphage. This high-quality, large-scale catalog of phage genomes will improve future virome studies and enable ecological and evolutionary analysis of human gut bacteriophages.
中文翻译:
人类肠道噬菌体多样性的大规模扩展
噬菌体通过创建促进生态适应的基因流网络来推动细菌群落的进化变化。然而,病毒多样性的程度及其在人类肠道中的流行程度仍然未知。在这里,我们介绍了肠道噬菌体数据库,该数据库包含 142,000 个非冗余病毒基因组(> 10 kb),通过挖掘包含 28,060 个全球分布的人类肠道宏基因组和 2,898 个培养肠道细菌参考基因组的数据集获得。宿主分配显示,厚壁菌门中的病毒多样性最高,并且约 36% 的病毒簇 (VC) 不限于单个物种,从而在系统发育不同的细菌物种之间创建基因流网络。流行病学分析发现,至少在 5 个大洲发现了 280 个全球分布的 VC,以及一个高度流行的噬菌体进化枝,其特征让人联想到 p-crAssphage。这种高质量、大规模的噬菌体基因组目录将改进未来的病毒组研究,并使人类肠道噬菌体的生态和进化分析成为可能。




















































 京公网安备 11010802027423号
京公网安备 11010802027423号