Bioorganic Chemistry ( IF 4.5 ) Pub Date : 2021-02-18 , DOI: 10.1016/j.bioorg.2021.104743 Xuemei Qin 1 , Leifu Yang 2 , Peng Liu 3 , Lifang Yang 4 , Linmeng Chen 4 , Liming Hu 2 , Mingguo Jiang 1
|
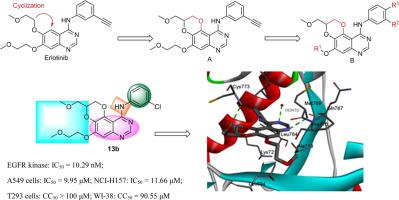
Epidermal growth factor receptor (EGFR) is the most attractive target for drug research in non-small cell lung cancer (NSCLC). The first-generation EGFR tyrosine kinase inhibitors (TKIs) Gefetinib and Elotinib showed good clinical efficacy on lung adenocarcinoma tumors, but almost all patients developed resistance to these inhibitors over time. Quinazoline and quinoline derivatives are common targeted inhibitors of EGFR kinase, and their structural optimization is an important direction for the development of effective targeted anticancer drugs. Based on these facts, a series of heterocyclic 2,3-dihydro-[1,4]dioxino[2,3-f]quinazoline derivatives have been designed and synthesized and their structures were confirmed by spectral analyses. The cytotoxic activity of the newly synthesized compounds was evaluated against the human kidney epithelial T293 cell line, normal lung cell lines WI-38, non-small cell lung cancer A549 and NCI-H157 cell lines using MTT. The tested compounds showed an evident anticancer activity against the tested cell lines, especially compound 13c, which was the most potent anticancer agent with half maximal inhibitory concentrations (IC50) between 8.82 and 10.24 μM. Docking study showed that compound 13b could be nicely bound to the ATP binding pocket of EGFR. In addition, the inhibitory activity of the target compounds against epidermal growth factor receptor tyrosine kinase (EGFR-TK) was evaluated. Results indicated the ability of the target compounds to inhibit EGFR-TK with half maximal inhibitory concentrations (IC50) in the range of 10.29 nM to 652.3 nM. In view of the reported compound activity, the structure deserves further optimization as cancer treatment agents.
中文翻译:
2,3-二氢-[1,4]二恶并[2,3-f]喹唑啉衍生物作为EGFR抑制剂的设计、合成和生物学评价
表皮生长因子受体(EGFR)是非小细胞肺癌(NSCLC)药物研究中最具吸引力的靶点。第一代EGFR酪氨酸激酶抑制剂(TKI)吉非替尼和埃洛替尼对肺腺癌肿瘤显示出良好的临床疗效,但随着时间的推移,几乎所有患者都对这些抑制剂产生了耐药性。喹唑啉和喹啉衍生物是常见的EGFR激酶靶向抑制剂,其结构优化是开发有效靶向抗癌药物的重要方向。基于这些事实,一系列杂环2,3-二氢-[1,4]二氧杂[2,3- f]喹唑啉衍生物已被设计和合成,其结构已通过光谱分析得到证实。使用 MTT 评估新合成化合物对人肾上皮 T293 细胞系、正常肺细胞系 WI-38、非小细胞肺癌 A549 和 NCI-H157 细胞系的细胞毒活性。受试化合物对受试细胞系显示出明显的抗癌活性,尤其是化合物13c,它是最有效的抗癌剂,半数抑制浓度 (IC 50 ) 介于 8.82 和 10.24 μM 之间。对接研究表明化合物13b可以很好地结合到 EGFR 的 ATP 结合口袋。此外,还评估了目标化合物对表皮生长因子受体酪氨酸激酶(EGFR-TK)的抑制活性。结果表明目标化合物具有抑制EGFR-TK的能力,半数抑制浓度(IC 50 )范围为10.29 nM至652.3 nM。鉴于报道的化合物活性,作为癌症治疗剂,该结构值得进一步优化。





























 京公网安备 11010802027423号
京公网安备 11010802027423号