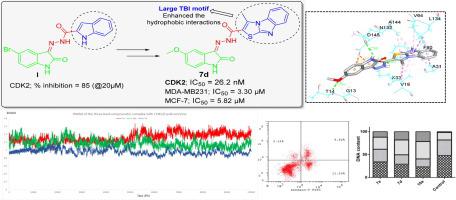
在当前的医学时代,人类健康正面临着诸多挑战,尤其是人类恶性肿瘤。因此,通过以选择性方式靶向肿瘤细胞的新疗法,将无情地增强这些恶性肿瘤的治疗手段。在这方面,目前的工作旨在开发一组新的小分子,其特征是通过可切割的酰肼接头(7a-e和10a-i)与噻唑并 [3,2- a ] 苯并咪唑 ( TBI ) 基序缀合的特权靛红支架。) 作为潜在的抗癌 CDK2 抑制剂。大三环TBI预计基序将在 CDK2 结合位点内实现大量疏水相互作用。大多数制备的杂合体在IC 50范围内显着抑制了两种检测细胞系的生长;(2.60 ± 1.47–20.90 ± 1.17 µM,针对 MDA-MB-231)和(1.27 ± 0.06–16.83 ± 0.95 µM,针对 MCF-7)。特别是,杂种7a、7d和10a对所检查的细胞系显示出强大的双重活性,因此被选中用于进一步研究。除了在 MDA-MB-231 和 MCF-7 细胞内诱导细胞凋亡外,它们还对细胞周期进程产生了显着的改变。此外,7a、7d和10a显示出有效的 CDK2 抑制作用(IC 50 = 96.46 ± 5.3、26.24 ± 1.4 和 42.95 ± 2.3 nM,分别)。正如预期的那样,对接模拟揭示了 TBI 环能够很好地适应并在 CDK2 结合位点的疏水口袋内建立几种疏水相互作用的能力。此外,对接模拟强调了在 H 键相互作用中掺入酰肼接头和未取代的靛红 (NH) 功能的重要性。有趣的是,在 100 ns MD 模拟中,最有效的 CDK2 抑制剂7d获得了最佳结合分数(-11.2 Kcal/mole)并与 CDK2 酶形成了最稳定的复合物(RMSD = 1.24 Å)。此外,MM-PBSA 计算将最低结合自由能归因于7d–CDK2 复合物 (-323.69 ± 15.17 kJ/mol)。这可归因于掺入了与关键 THR14 氨基酸残基形成额外氢键的 5-OCH 3基团。最后,这些结果表明杂交7d作为有前景的乳腺癌抗肿瘤剂和 CDK2 抑制剂是进一步优化的良好候选者。
 "点击查看英文标题和摘要"
"点击查看英文标题和摘要"
Development of isatin-thiazolo[3,2-a]benzimidazole hybrids as novel CDK2 inhibitors with potent in vitro apoptotic anti-proliferative activity: Synthesis, biological and molecular dynamics investigations
In the current medical era, human health is experiencing numerous challenges, particularly the human malignancies. Therefore, the therapeutic arsenal for these malignancies is to be inexorably enhanced with new treatments that target tumor cells in a selective manner. In this regard, the present work aims at developing a new set of small molecules featuring the privileged isatin scaffold conjugated with a thiazolo[3,2-a]benzimidazole (TBI) motif through a cleavable hydrazide linker (7a-e and 10a-i) as potential anticancer CDK2 inhibitors. The large tricyclic TBI motif is anticipated to achieve a plethora of hydrophobic interactions within the CDK2 binding site. The growth of the two examined cell lines was significantly inhibited by most the prepared hybrids with IC50 ranges; (2.60 ± 1.47–20.90 ± 1.17 µM, against MDA-MB-231) and (1.27 ± 0.06–16.83 ± 0.95 µM, against MCF-7). In particular, hybrids 7a, 7d and 10a displayed potent dual activity against the examined cell lines, and thus selected for further investigations. They exerted a significance alteration in the cell cycle progression, in addition to an apoptosis induction within both MDA-MB-231 and MCF-7 cells. Furthermore, 7a, 7d and 10a displayed potent CDK2 inhibitory action (IC50 = 96.46 ± 5.3, 26.24 ± 1.4 and 42.95 ± 2.3 nM, respectively). The docking simulations unveiled, as expected, the ability of the TBI ring to well-accommodate and establish several hydrophobic interactions within a hydrophobic pocket in the CDK2 binding site. Also, the docking simulations highlighted the significance of incorporation of the hydrazide linker and isatin unsubstituted (NH) functionality in the H-bonding interactions. Interestingly, the most potent CDK2 inhibitor 7d achieved the best binding score (-11.2 Kcal/mole) and formed the most stable complex with CDK2 enzyme (RMSD = 1.24 Å) in a 100 ns MD simulation. In addition, the MM-PBSA calculations ascribed the lowest binding free energy to the 7d–CDK2 complex (−323.69 ± 15.17 kJ/mol). This could be attributed to an incorporation of the 5-OCH3 group that was engaged in an extra hydrogen bonding with key THR14 amino acid residue. Finally, these results suggested hybrid 7d as a good candidate for further optimization as promising breast cancer antitumor agent and CDK2 inhibitor.
































 京公网安备 11010802027423号
京公网安备 11010802027423号