当前位置:
X-MOL 学术
›
ACS Catal.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
On the Reaction Mechanism of Direct H2O2 Formation over Pd Catalysts
ACS Catalysis ( IF 11.3 ) Pub Date : 2021-02-12 , DOI: 10.1021/acscatal.0c05548 Lin Chen 1 , J. Will Medlin 2 , Henrik Grönbeck 1
ACS Catalysis ( IF 11.3 ) Pub Date : 2021-02-12 , DOI: 10.1021/acscatal.0c05548 Lin Chen 1 , J. Will Medlin 2 , Henrik Grönbeck 1
Affiliation
|
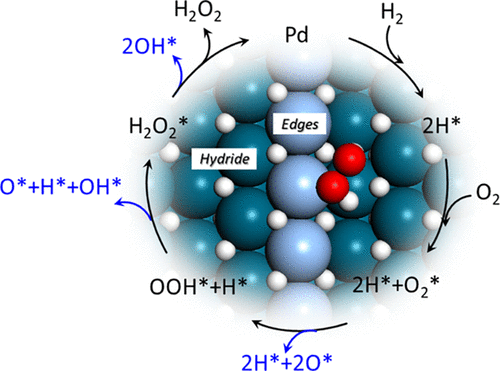
Hydrogen peroxide (H2O2) is an effective green oxidant, which is used in many industrial processes. Here, the reaction mechanism for direct formation of H2O2 from H2 and O2 over Pd catalysts is studied using density functional theory calculations and mean-field kinetic modeling. The state of the catalyst as a function of reaction conditions is determined from ab initio thermodynamics. It is found that Pd is in a hydride phase during typical reaction conditions. Reaction landscapes are constructed for the reaction over PdH(111) and PdH(211). Formation of H2O2 instead of H2O requires that O2 adsorbs and that the surface intermediates O2, OOH, and H2O2 do not dissociate. We find that these requirements are fulfilled on the stepped PdH(211) surface. Surface steps are needed for O2 chemisorption as the adsorption on PdH(111) is endothermic. The high H coverage on the surface of the hydride is important to slow down the unwanted scission of the O–O bond and promote the desorption of the products. Comparative calculations for the Pd(111) surface show that this surface is inactive for both H2O2 and H2O formation below room temperature for typical reaction mixtures. Our findings demonstrate the importance of surface steps and high hydrogen coverage for direct synthesis of H2O2 from H2 and O2 over Pd catalysts. The results imply that the selectivity of the reaction toward H2O2 is enhanced by a high partial pressure of H2, which is in agreement with experimental observations.
中文翻译:

Pd催化剂上直接形成H 2 O 2的反应机理
过氧化氢(H 2 O 2)是一种有效的绿色氧化剂,已在许多工业过程中使用。在此,使用密度泛函理论计算和平均场动力学模型研究了在Pd催化剂上由H 2和O 2直接形成H 2 O 2的反应机理。催化剂的状态作为反应条件的函数是从头算热力学确定的。发现在典型的反应条件下Pd处于氢化物相。构建了用于PdH(111)和PdH(211)上反应的反应态势。形成H 2 O 2代替H 2O需要O 2吸附并且表面中间体O 2,OOH和H 2 O 2不会解离。我们发现在阶梯式PdH(211)表面上满足了这些要求。O 2化学吸附需要表面步骤,因为在PdH(111)上的吸附是吸热的。氢化物表面的高H覆盖率对于减缓O-O键的不必要的断裂并促进产物的解吸非常重要。对Pd(111)表面的比较计算表明,该表面对H 2 O 2和H 2均不起作用对于典型的反应混合物,在室温以下会形成O。我们的发现证明了在Pd催化剂上由H 2和O 2直接合成H 2 O 2的表面步骤和高氢覆盖率的重要性。该结果意味着,朝向小时后,反应的选择性2 Ó 2通过H的分压高增强2,这是在与实验观察一致。
更新日期:2021-03-05
中文翻译:
Pd催化剂上直接形成H 2 O 2的反应机理
过氧化氢(H 2 O 2)是一种有效的绿色氧化剂,已在许多工业过程中使用。在此,使用密度泛函理论计算和平均场动力学模型研究了在Pd催化剂上由H 2和O 2直接形成H 2 O 2的反应机理。催化剂的状态作为反应条件的函数是从头算热力学确定的。发现在典型的反应条件下Pd处于氢化物相。构建了用于PdH(111)和PdH(211)上反应的反应态势。形成H 2 O 2代替H 2O需要O 2吸附并且表面中间体O 2,OOH和H 2 O 2不会解离。我们发现在阶梯式PdH(211)表面上满足了这些要求。O 2化学吸附需要表面步骤,因为在PdH(111)上的吸附是吸热的。氢化物表面的高H覆盖率对于减缓O-O键的不必要的断裂并促进产物的解吸非常重要。对Pd(111)表面的比较计算表明,该表面对H 2 O 2和H 2均不起作用对于典型的反应混合物,在室温以下会形成O。我们的发现证明了在Pd催化剂上由H 2和O 2直接合成H 2 O 2的表面步骤和高氢覆盖率的重要性。该结果意味着,朝向小时后,反应的选择性2 Ó 2通过H的分压高增强2,这是在与实验观察一致。
































 京公网安备 11010802027423号
京公网安备 11010802027423号