当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Raman Spectra of Persistent Radical Anions from Benzophenone, Fluorenone, 2,2′-Bipyridyl, 4,4′-Di-tert-butyl-2,2′-dipyridyl, and Anthracene: Excellent Agreement between DFT and Experiment for Highly Delocalized Radical Systems
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2021-02-05 , DOI: 10.1021/acs.jpcb.0c04742 Antoine Juneau 1 , Mathieu Frenette 1
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2021-02-05 , DOI: 10.1021/acs.jpcb.0c04742 Antoine Juneau 1 , Mathieu Frenette 1
Affiliation
|
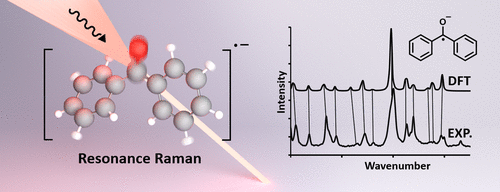
We report detailed Raman spectra for the neutral and radical anion forms of benzophenone, fluorenone, 2,2′-bipyridyl, 4,4′-di-tert-butyl-2,2′-dipyridyl, and anthracene. Density functional theory (DFT) predictions for the Raman spectra of these molecules give additional insight into the assignment of each vibrational mode. While the use of DFT has been problematic in quantifying the thermochemistry of highly delocalized radicals, we find that DFT-predicted spectra using the popular B3LYP functional are in excellent agreement with the observed Raman spectra. In the case of the two bipyridyl compounds, the Raman spectra allowed us to conclude that the cis form of the radical anion complexed to a sodium cation was the preferred configuration. Benzophenone and fluorenone radical anions gave a significantly weakened C═O bond stretching vibrational frequency as expected from the population of an antibonding π* orbital. For benzophenone, the C═O vibration dropped from 1659 to 1403 cm–1 upon reduction. Similarly, fluorenone showed a C═O vibration observed at 1719 cm–1 for the neutral form that decreased to 1522 cm–1 for the radical anion. The structurally rigid anthracene showed relatively smaller Raman band shifts upon single-electron reduction as the π* orbital is more equally delocalized on the entire structure. In total, we correlated 65 DFT-predicted vibrational modes for the neutral molecules with an overall error of 7.1 cm–1 (root-mean-square errors (RMSEs)) and 67 DFT-predicted vibrational modes for radical anions with an overall error of 9.9 cm–1. These comparisons between theory and experiment are another example to demonstrate the power of DFT in predicting the identity and geometry of molecules using Raman spectroscopy.
中文翻译:

苯甲酮,芴酮,2,2'-联吡啶,4,4'-二叔丁基-2,2'-联吡啶和蒽的持久性自由基阴离子的拉曼光谱:DFT与高度离域自由基体系实验之间的优异一致性
我们报告中详细拉曼光谱的中性和阴离子自由基形式的二苯甲酮,芴,2,2'-联吡啶,4,4'-二-叔-丁基-2,2'-二吡啶基和蒽。这些分子的拉曼光谱的密度泛函理论(DFT)预测可进一步了解每种振动模式的分配。虽然使用DFT在量化高度离域的自由基的热化学方面一直存在问题,但我们发现使用流行的B3LYP官能团进行DFT预测的光谱与观察到的拉曼光谱非常吻合。在两种联吡啶化合物的情况下,拉曼光谱使我们得出结论,与钠阳离子络合的自由基阴离子的顺式是优选的构型。苯甲酮和芴酮自由基阴离子显着减弱了C═O键的拉伸振动频率,这是由反键π*轨道的数量所预期的。对于二苯甲酮,C═O振动从1659降至1403 cm还原时为–1。同样,芴酮在中性形式下在1719 cm –1处观察到C = O振动,而在自由基阴离子上下降到1522 cm –1。具有结构刚性的蒽在单电子还原时显示出相对较小的拉曼带位移,因为π*轨道在整个结构上的分布更均等。总共,我们将中性分子的65种DFT预测的振动模式与总误差为7.1 cm –1(均方根误差(RMSEs))相关,将自由基阴离子的67种DFT预测的振动模式与总误差相关联。 9.9厘米–1。理论与实验之间的这些比较是另一个例子,证明了DFT在使用拉曼光谱法预测分子的同一性和几何形状方面的强大能力。
更新日期:2021-02-18
中文翻译:
苯甲酮,芴酮,2,2'-联吡啶,4,4'-二叔丁基-2,2'-联吡啶和蒽的持久性自由基阴离子的拉曼光谱:DFT与高度离域自由基体系实验之间的优异一致性
我们报告中详细拉曼光谱的中性和阴离子自由基形式的二苯甲酮,芴,2,2'-联吡啶,4,4'-二-叔-丁基-2,2'-二吡啶基和蒽。这些分子的拉曼光谱的密度泛函理论(DFT)预测可进一步了解每种振动模式的分配。虽然使用DFT在量化高度离域的自由基的热化学方面一直存在问题,但我们发现使用流行的B3LYP官能团进行DFT预测的光谱与观察到的拉曼光谱非常吻合。在两种联吡啶化合物的情况下,拉曼光谱使我们得出结论,与钠阳离子络合的自由基阴离子的顺式是优选的构型。苯甲酮和芴酮自由基阴离子显着减弱了C═O键的拉伸振动频率,这是由反键π*轨道的数量所预期的。对于二苯甲酮,C═O振动从1659降至1403 cm还原时为–1。同样,芴酮在中性形式下在1719 cm –1处观察到C = O振动,而在自由基阴离子上下降到1522 cm –1。具有结构刚性的蒽在单电子还原时显示出相对较小的拉曼带位移,因为π*轨道在整个结构上的分布更均等。总共,我们将中性分子的65种DFT预测的振动模式与总误差为7.1 cm –1(均方根误差(RMSEs))相关,将自由基阴离子的67种DFT预测的振动模式与总误差相关联。 9.9厘米–1。理论与实验之间的这些比较是另一个例子,证明了DFT在使用拉曼光谱法预测分子的同一性和几何形状方面的强大能力。




















































 京公网安备 11010802027423号
京公网安备 11010802027423号