Journal of Molecular Liquids ( IF 5.3 ) Pub Date : 2021-01-23 , DOI: 10.1016/j.molliq.2021.115449 I.V. Fedorova , M.A. Krestyaninov , L.P. Safonova
|
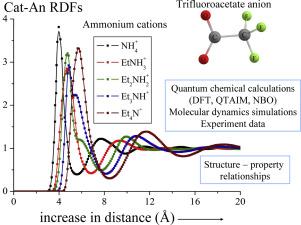
Ionic liquids (ILs) raise quite interesting but complicated questions for computational study due to the fact that different interactions, e.g., electrostatic interactions, charge-transfer interactions, steric repulsion, dispersion and hydrogen bonding that determine structural organization of ILs, need to be described properly. In this work, we present a comprehensive understanding of structural properties and interactions between ions in trifluoroacetate-based ILs with ammonium cations containing ethyl groups with the general formula EtnNH4-n (n = 0–4). This study includes both gas-phase and implicit solvent calculations of single ionic pairs (DFT, QTAIM and NBO methods) and molecular dynamics (MD) simulations of the ILs themselves. Based on the gas-phase calculations we show that the ionic and molecular pairs with very strong hydrogen bonds are formed by diethylamine and triethylamine with trifluoroacetic acid, while only molecular pairs are generated from ammonia and ethylamine. Unlike this, the use of implicit solvent calculations for all compounds leads to the result that formation of ionic pairs becomes energetically more favorable which, in turn, improves the agreement with experiment. The trend in geometric parameters of hydrogen bonds obtained from MD simulations of the ILs is similar to that which is observed for the implicit solvent calculations of the ion pairs. The increase in the number of alkyl groups in the cation would decrease interaction between the ions as in the ionic pair, as well in the ionic liquid itself. Moreover, we suggest that the change in the ion-ion interaction is one of the most important factors that affects the melting points variation of the ILs for this series.
中文翻译:
基于三氟乙酸盐的离子液体中的结构和离子-离子相互作用:量子化学和分子动力学模拟研究
由于需要描述决定离子液体结构的不同相互作用,例如静电相互作用,电荷转移相互作用,空间排斥,分散和氢键结合,离子液体(ILs)引起了相当有趣但复杂的计算研究问题。正确地。在这项工作中,我们对基于三氟乙酸酯的ILs中的离子与通式为Et n NH 4-n(n = 0–4)。这项研究包括单个离子对的气相和隐式溶剂计算(DFT,QTAIM和NBO方法)以及IL自身的分子动力学(MD)模拟。基于气相计算,我们表明具有非常强氢键的离子对和分子对是由二乙胺和三乙胺与三氟乙酸形成的,而只有分子对是由氨和乙胺生成的。与此不同,对所有化合物使用隐式溶剂计算会导致离子对的形成在能量上更加有利的结果,从而改善了与实验的一致性。从IL的MD模拟获得的氢键几何参数的趋势与对离子对的隐式溶剂计算所观察到的趋势相似。阳离子中烷基数目的增加将减少离子之间的相互作用,如在离子对中,以及在离子液体本身中。此外,我们建议离子-离子相互作用的变化是影响此系列IL熔点变化的最重要因素之一。










































 京公网安备 11010802027423号
京公网安备 11010802027423号