Bioorganic & Medicinal Chemistry ( IF 3.3 ) Pub Date : 2021-01-23 , DOI: 10.1016/j.bmc.2021.116039 Tsutomu Fukuda 1 , Mizuho Anzai 2 , Akane Nakahara 2 , Kentaro Yamashita 2 , Kazuaki Matsukura 3 , Fumito Ishibashi 3 , Yusuke Oku 4 , Naoyuki Nishiya 4 , Yoshimasa Uehara 4 , Masatomo Iwao 2
|
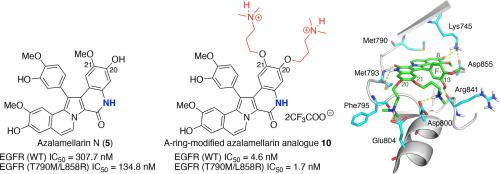
Azalamellarin N, a synthetic lactam congener of the marine natural product lamellarin N, and its A-ring-modified analogues were synthesized and evaluated as potent and non-covalent inhibitors of the drug-resistant epidermal growth factor receptor T790M/L858R mutant. An in vitro tyrosine kinase assay indicated that the inhibitory activities of the synthetic azalamellarin analogues were higher than those of the corresponding lamellarins. The azalamellarin analogue bearing two 3-(dimethylamino)propoxy groups at C20- and C21-positions exhibited the highest activity and selectivity against the mutant kinase [IC50 (T790M/L858R) = 1.7 nM; IC50 (WT) = 4.6 nM]. The inhibitory activity was attributed to the hydrogen bonding interaction between the lactam NH group of the B-ring and carbonyl group of a methionine residue.
中文翻译:
作为 EGFR T790M/L858R 突变体的非共价抑制剂的氮芥素 N 及其 A 环修饰类似物的合成和评价
Azalamellarin N,一种海洋天然产物 lamellarin N 的合成内酰胺同源物,及其 A 环修饰的类似物被合成并评估为耐药表皮生长因子受体 T790M/L858R 突变体的有效和非共价抑制剂。体外酪氨酸激酶试验表明,合成的氮杂紫杉醇类似物的抑制活性高于相应的紫胨。在 C20 和 C21 位带有两个 3-(二甲氨基)丙氧基的氮杂紫杉醇类似物对突变激酶表现出最高的活性和选择性 [IC 50 (T790M/L858R) = 1.7 nM;集成电路50(WT) = 4.6 nM]。抑制活性归因于B环的内酰胺NH基团和甲硫氨酸残基的羰基之间的氢键相互作用。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号