当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Adsorption of Normal-Alkanes on Fe(110), FeO(110), and Fe2O3(0001): Influence of Iron Oxide Surfaces
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2015-05-27 00:00:00 , DOI: 10.1021/acs.jpcc.5b01847
Thi D. Ta 1 , A. Kiet Tieu 1 , Hongtao Zhu 1 , Buyung Kosasih 1
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2015-05-27 00:00:00 , DOI: 10.1021/acs.jpcc.5b01847
Thi D. Ta 1 , A. Kiet Tieu 1 , Hongtao Zhu 1 , Buyung Kosasih 1
Affiliation
|
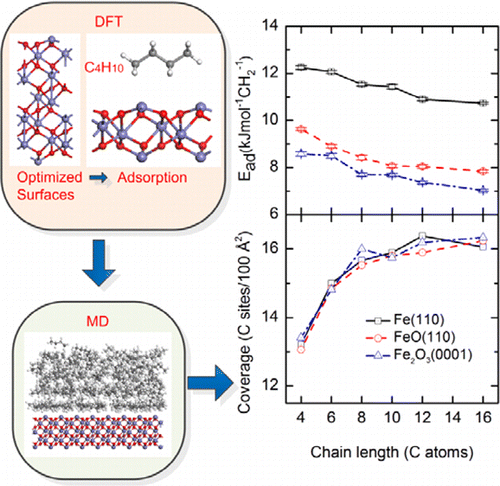
A comparative analysis of adsorption of six normal-alkanes (CNH2N+2, N = 4, 6, 8, 10, 12, 16) on Fe(110), FeO(110), and Fe2O3(0001) was carried out using classical molecular dynamics (MD) simulation. A realistic model system for adsorbed alkanes was employed using the COMPASS force field (FF), while the appropriate relaxed surfaces and an effective interfacial potential were obtained from ab initio calculations. The results show that butane molecules orient randomly on Fe(110) and Fe2O3(0001) surfaces, but they preferentially orient in the (010) direction on FeO(110) at low temperature. Additionally, alkanes adsorb physically on Fe(110), FeO(110), and Fe2O3(0001), in the following decreasing order Fe(110) > FeO(110) > Fe2O3(0001). The adsorption energies per saturated carbon site decrease with an increase of molecular chain length, and this propensity is similar for different surface potentials. In contrast, the saturated carbon density is insensitive to the surface potentials and shows an increasing trend for short alkane chains, but it remains steady for longer chains.
中文翻译:

正烷烃在Fe(110),FeO(110)和Fe 2 O 3(0001)上的吸附:氧化铁表面的影响
对Fe(110),FeO(110)和Fe 2 O 3(C N H 2 N +2,N = 4、6、8、10、12、16)六种正构烷烃的吸附比较分析0001)是使用经典分子动力学(MD)模拟进行的。使用COMPASS力场(FF),采用了吸附烷烃的逼真的模型系统,而适当的松弛表面和有效的界面电势是从头算得到的。结果表明,丁烷分子在Fe(110)和Fe 2 O 3上随机取向(0001)表面,但它们在低温下优先在FeO(110)上沿(010)方向取向。此外,烷烃以下列递减顺序依次物理吸附在Fe(110),FeO(110)和Fe 2 O 3(0001)上:Fe(110)> FeO(110)> Fe 2 O 3(0001)。每个饱和碳位的吸附能随着分子链长度的增加而降低,并且这种倾向对于不同的表面电势是相似的。相反,饱和碳密度对表面电势不敏感,并且对于短链烷烃链显示出增加的趋势,但是对于较长链烷烃则保持稳定。
更新日期:2015-05-27
中文翻译:
正烷烃在Fe(110),FeO(110)和Fe 2 O 3(0001)上的吸附:氧化铁表面的影响
对Fe(110),FeO(110)和Fe 2 O 3(C N H 2 N +2,N = 4、6、8、10、12、16)六种正构烷烃的吸附比较分析0001)是使用经典分子动力学(MD)模拟进行的。使用COMPASS力场(FF),采用了吸附烷烃的逼真的模型系统,而适当的松弛表面和有效的界面电势是从头算得到的。结果表明,丁烷分子在Fe(110)和Fe 2 O 3上随机取向(0001)表面,但它们在低温下优先在FeO(110)上沿(010)方向取向。此外,烷烃以下列递减顺序依次物理吸附在Fe(110),FeO(110)和Fe 2 O 3(0001)上:Fe(110)> FeO(110)> Fe 2 O 3(0001)。每个饱和碳位的吸附能随着分子链长度的增加而降低,并且这种倾向对于不同的表面电势是相似的。相反,饱和碳密度对表面电势不敏感,并且对于短链烷烃链显示出增加的趋势,但是对于较长链烷烃则保持稳定。
































 京公网安备 11010802027423号
京公网安备 11010802027423号