Journal of Molecular Modeling ( IF 2.1 ) Pub Date : 2021-01-05 , DOI: 10.1007/s00894-020-04619-7 Rao Aqil Shehzad , Shabbir Muhammad , Javed Iqbal , Abdullah G. Al-Sehemi , Muhammad Yaseen , Zouhaier Aloui , Muhammad Khalid
|
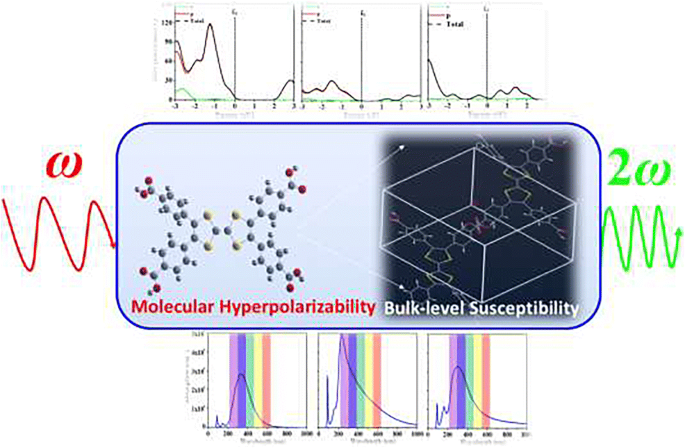
In the present investigation, we use a dual computational approach (at single molecular and solid-state levels) to explore the optoelectronic and nonlinear optical (NLO) properties of cross-shaped derivatives. The solid-state electronic band structures of the compounds 1–3 (the derivatives of tetracarboxylic acid in cross-shaped having the core of benzene (1), pyrazinoquinoxaline (2), and tetrathiafulvalene (3)) are calculated. The calculated band gaps for compounds 1–2 are found to be direct bad gaps and compound 3 to be indirect bad gap with energy gaps of 2.749, 1.765, and 0.875 eV, respectively. The important optical properties including refractive index, absorption coefficients, loss functions, and extinction coefficient of these semiconductors are calculated at bulk level to seek their potential applications as efficient optoelectronic materials. Additionally, we use the Lorentz approximation to calculate the third-order NLO susceptibilities of compounds 1–3 using the molecular hyperpolarizability and solid-state parameters. The calculated third-order NLO susceptibilities of compounds 1–3 are found to be 6.92 × 10−12, 64.0 × 10−12, and 26.3 × 10−12 esu, respectively. Thus, the present study not only provides a way to connect the calculated third-order molecular NLO polarizability to NLO susceptibilities for compounds 1–3 through Lorentz approximation but also highlights the importance of central core modifications on their NLO susceptibilities.
Graphical abstract
中文翻译:
探索十字形分子的光电和三阶非线性光学磁化率:从分子到材料水平的见解
在本研究中,我们使用双重计算方法(在单个分子和固态水平上)来探索十字形导数的光电和非线性光学(NLO)特性。固态电子带的化合物的结构1 - 3(在四羧酸的衍生物十字形具有苯(核心1),pyrazinoquinoxaline(2),和四硫富瓦(3))被计算。化合物1 – 2的计算带隙被发现是直接的坏间隙,化合物3分别为2.749、1.765和0.875 eV的间接能隙。这些半导体的重要光学特性(包括折射率,吸收系数,损耗函数和消光系数)是按体积计算的,以寻求其作为有效光电材料的潜在应用。此外,我们使用洛伦兹近似来计算化合物的三阶非线性光学敏感性1 - 3使用分子超极化率和固态参数。化合物的计算三阶非线性光学敏感性1 - 3被发现是6.92×10 -12,64.0×10 -12,和26.3×10 -12 分别。因此,本研究不仅提供了一种方法所计算出的三阶分子NLO极化连接到NLO磁化率为化合物1 - 3通过洛伦兹近似而且还突出在其NLO敏感性中央芯修饰的重要性。






























 京公网安备 11010802027423号
京公网安备 11010802027423号