当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
General Theory of Fragment Linking in Molecular Design: Why Fragment Linking Rarely Succeeds and How to Improve Outcomes
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-12-29 , DOI: 10.1021/acs.jctc.0c01004 Haoyu S. Yu 1 , Kalyan Modugula 2 , Osamu Ichihara 3 , Kimberly Kramschuster 4 , Simon Keng 4 , Robert Abel 1 , Lingle Wang 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-12-29 , DOI: 10.1021/acs.jctc.0c01004 Haoyu S. Yu 1 , Kalyan Modugula 2 , Osamu Ichihara 3 , Kimberly Kramschuster 4 , Simon Keng 4 , Robert Abel 1 , Lingle Wang 1
Affiliation
|
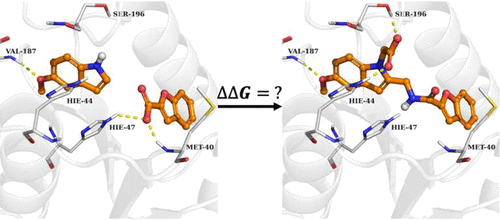
Linking two fragments binding in nearby subpockets together has become an important technique in fragment-based drug discovery to optimize the binding potency of fragment hits. Despite the expected favorable translational and orientational entropic contribution to the binding free energy of the linked molecule, brute force enumeration of chemical linker for linking fragments is rarely successful, and the vast majority of linked molecules do not exhibit the expected gains of binding potency. In this paper, we examine the physical factors that contribute to the change of binding free energy from fragment linking and develop a method to rigorously calculate these different physical contributions. We find from these analyses that multiple confounding factors make successful fragment linking strategies rare, including (1) possible change of the binding mode of the fragments in the linked state compared to separate binding of the fragments, (2) unfavorable intramolecular strain energy of the bioactive conformation of the linked molecule, (3) unfavorable interaction between the linker and the protein, (4) favorable interaction energies between two fragments in solution when not chemically linked that offset the expected entropy loss for the formation of fragment pair, (5) complex compensating configurational entropic effects beyond the simplistic rotational and translational analysis. We here have applied a statistically mechanically rigorous approach to compute the fragment linking coefficients of 10 pharmaceutically interesting systems and quantify the contribution of each physical component to the binding free energy of the linked molecule. Based on these studies, we have found that the change in the relative configurational entropy of the two fragments in the protein binding pocket (a term neglected to our knowledge in all previous analyses) substantially offsets the favorable expected rotational and translational entropic contributions to the binding free energy of the linked molecule. This configurational restriction of the fragments in the binding pocket of the proteins is found to be, in our analysis, the dominant reason why most fragment linking strategies do not exhibit the expected gains of binding potency. These findings have further provided rich physical insights, which we expect should facilitate more successful fragment linking strategies to be formulated in the future.
中文翻译:

分子设计中片段连接的一般理论:为什么片段连接很少成功以及如何改善结果
将结合在附近子口袋中的两个片段连接在一起已成为基于片段的药物发现中优化片段命中的结合力的一项重要技术。尽管预期对连接分子的结合自由能具有有利的平移和取向熵的贡献,但是用于连接片段的化学连接物的蛮力枚举很少成功,并且绝大多数连接分子没有表现出预期的结合力。在本文中,我们研究了影响片段连接中结合自由能变化的物理因素,并开发了一种方法来严格计算这些不同的物理贡献。从这些分析中我们发现,多种混杂因素使成功的片段连接策略稀有,包括(1)与片段的单独结合相比,处于连接状态的片段的结合模式可能发生变化;(2)所链接分子的生物活性构象的不利的分子内应变能;(3)接头与分子之间的不利相互作用蛋白质;(4)未化学连接时溶液中两个片段之间的良好相互作用能,抵消了片段对形成所需的熵损失;(5)除了简单的旋转和平移分析外,复杂的结构熵效应得到补偿。我们在这里应用了统计上严格的机械方法来计算10个药学上有意义的系统的片段连接系数,并量化每种物理成分对连接分子的结合自由能的贡献。基于这些研究,我们发现蛋白质结合口袋中两个片段的相对构型熵的变化(一个在所有先前分析中都被忽略的术语)大大抵消了对结合的有利的预期旋转和翻译熵连接分子的自由能。在我们的分析中,发现蛋白质结合口袋中片段的这种构型限制是大多数片段连接策略未显示出预期的结合力增益的主要原因。这些发现进一步提供了丰富的物理见解,我们希望这些见解将有助于将来制定更成功的片段连接策略。我们已经发现,蛋白质结合口袋中两个片段的相对构型熵的改变(一个在所有先前分析中都被我们忽略的术语)基本上抵消了对连接的结合自由能的有利的预期旋转和翻译熵的贡献。分子。在我们的分析中,发现蛋白质结合口袋中片段的这种构型限制是大多数片段连接策略未显示出预期的结合力增益的主要原因。这些发现进一步提供了丰富的物理见解,我们希望这些见解将有助于将来制定更成功的片段连接策略。我们已经发现,蛋白质结合口袋中两个片段的相对构型熵的改变(一个在所有先前分析中都被我们忽略的术语)基本上抵消了对连接的结合自由能的有利的预期旋转和翻译熵的贡献。分子。在我们的分析中,发现蛋白质结合口袋中片段的这种构型限制是大多数片段连接策略未显示出预期的结合力增益的主要原因。这些发现进一步提供了丰富的物理见解,我们希望这些见解将有助于将来制定更成功的片段连接策略。
更新日期:2020-12-29
中文翻译:
分子设计中片段连接的一般理论:为什么片段连接很少成功以及如何改善结果
将结合在附近子口袋中的两个片段连接在一起已成为基于片段的药物发现中优化片段命中的结合力的一项重要技术。尽管预期对连接分子的结合自由能具有有利的平移和取向熵的贡献,但是用于连接片段的化学连接物的蛮力枚举很少成功,并且绝大多数连接分子没有表现出预期的结合力。在本文中,我们研究了影响片段连接中结合自由能变化的物理因素,并开发了一种方法来严格计算这些不同的物理贡献。从这些分析中我们发现,多种混杂因素使成功的片段连接策略稀有,包括(1)与片段的单独结合相比,处于连接状态的片段的结合模式可能发生变化;(2)所链接分子的生物活性构象的不利的分子内应变能;(3)接头与分子之间的不利相互作用蛋白质;(4)未化学连接时溶液中两个片段之间的良好相互作用能,抵消了片段对形成所需的熵损失;(5)除了简单的旋转和平移分析外,复杂的结构熵效应得到补偿。我们在这里应用了统计上严格的机械方法来计算10个药学上有意义的系统的片段连接系数,并量化每种物理成分对连接分子的结合自由能的贡献。基于这些研究,我们发现蛋白质结合口袋中两个片段的相对构型熵的变化(一个在所有先前分析中都被忽略的术语)大大抵消了对结合的有利的预期旋转和翻译熵连接分子的自由能。在我们的分析中,发现蛋白质结合口袋中片段的这种构型限制是大多数片段连接策略未显示出预期的结合力增益的主要原因。这些发现进一步提供了丰富的物理见解,我们希望这些见解将有助于将来制定更成功的片段连接策略。我们已经发现,蛋白质结合口袋中两个片段的相对构型熵的改变(一个在所有先前分析中都被我们忽略的术语)基本上抵消了对连接的结合自由能的有利的预期旋转和翻译熵的贡献。分子。在我们的分析中,发现蛋白质结合口袋中片段的这种构型限制是大多数片段连接策略未显示出预期的结合力增益的主要原因。这些发现进一步提供了丰富的物理见解,我们希望这些见解将有助于将来制定更成功的片段连接策略。我们已经发现,蛋白质结合口袋中两个片段的相对构型熵的改变(一个在所有先前分析中都被我们忽略的术语)基本上抵消了对连接的结合自由能的有利的预期旋转和翻译熵的贡献。分子。在我们的分析中,发现蛋白质结合口袋中片段的这种构型限制是大多数片段连接策略未显示出预期的结合力增益的主要原因。这些发现进一步提供了丰富的物理见解,我们希望这些见解将有助于将来制定更成功的片段连接策略。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号