当前位置:
X-MOL 学术
›
Acc. Chem. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Branching Out: Rhodium-Catalyzed Allylation with Alkynes and Allenes
Accounts of Chemical Research ( IF 16.4 ) Pub Date : 2016-07-25 00:00:00 , DOI: 10.1021/acs.accounts.6b00252 Philipp Koschker 1 , Bernhard Breit 1
Accounts of Chemical Research ( IF 16.4 ) Pub Date : 2016-07-25 00:00:00 , DOI: 10.1021/acs.accounts.6b00252 Philipp Koschker 1 , Bernhard Breit 1
Affiliation
|
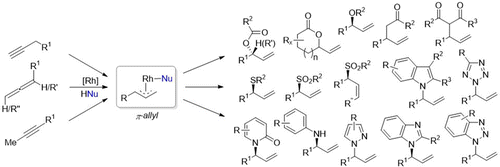
We present a new and efficient strategy for the atom-economic transformation of both alkynes and allenes to allylic functionalized structures via a Rh-catalyzed isomerization/addition reaction which has been developed in our working group. Our methodology thus grants access to an important structural class valued in modern organic chemistry for both its versatility for further functionalization and the potential for asymmetric synthesis with the construction of a new stereogenic center. This new methodology, inspired by mechanistic investigations by Werner in the late 1980s and based on preliminary work by Yamamoto and Trost, offers an attractive alternative to other established methods for allylic functionalization such as allylic substitution or allylic oxidation. The main advantage of our methodology consists of the inherent atom economy in comparison to allylic oxidation or substitution, which both produce stoichiometric amounts of waste and, in case of the substitution reaction, require prefunctionalization of the starting material. Starting out with the discovery of a highly branched-selective coupling reaction of carboxylic acids with terminal alkynes using a Rh(I)/DPEphos complex as the catalyst system, over the past 5 years we were able to continuously expand upon this chemistry, introducing various (pro)nucleophiles for the selective C–O, C–S, C–N, and C–C functionalization of both alkynes and the double-bond isomeric allenes by choosing the appropriate rhodium/bidentate phosphine catalyst. Thus, valuable compounds such as branched allylic ethers, sulfones, amines, or γ,δ-unsaturated ketones were successfully synthesized in high yields and with a broad substrate scope. Beyond the branched selectivity inherent to rhodium, many of the presented methodologies display additional degrees of selectivity in regard to regio-, diastereo-, and enantioselective transformations, with one example even proceeding via a dynamic kinetic resolution. Many advances presented in this account were driven by detailed mechanistic investigations including DFT-calculations, ESI-MS and in situ IR experiments and enabled the application of our chemistry for target-oriented syntheses demonstrated by several examples shown herein. In general, this research topic has matured over the past years into a viable option when synthesizing chiral compounds, from small molecules such as quercus lactones to complex target structures such as Homolargazole or Clavosolide A. This demonstrates the importance and utility of these coupling reactions, especially considering the ease with which carbon–heteroatom bonds can be built stereoselectively, with many of the product classes displaying motifs common in modern APIs.
中文翻译:

分支:铑催化的炔烃和丙二烯的烯丙基化
我们提出了一种新的有效策略,通过我们的工作组开发的一种通过Rh催化的异构化/加成反应,将炔烃和丙二烯进行原子经济转化为烯丙基官能化结构的方法。因此,我们的方法学使人们能够利用现代有机化学中有价值的重要结构类别,因为它具有进一步实现功能化的多功能性以及通过构建新的立体异构中心进行不对称合成的潜力。这种新方法是受到1980年代后期Werner进行的机械研究的启发,并基于Yamamoto和Trost的初步工作,为其他已建立的烯丙基功能化方法(如烯丙基取代或烯丙基氧化)提供了有吸引力的替代方法。与烯丙基氧化或取代相比,我们方法的主要优势在于固有的原子经济性,这既会产生化学计量的废物,而且在发生取代反应的情况下,还需要对原料进行预功能化。从发现使用Rh(I)/ DPEphos络合物作为催化剂体系发现羧酸与末端炔烃的高度支化-选择性偶联反应开始,在过去的5年中,我们能够不断扩展这种化学反应,引入各种(亲)亲核试剂通过选择合适的铑/双齿膦催化剂对炔烃和双键异构艾伦进行选择性C–O,C–S,C–N和C–C功能化。因此,有价值的化合物,例如支链烯丙基醚,砜,胺或γ,δ-不饱和酮已成功地以高收率和广泛的底物范围合成。除了铑固有的支化选择性外,许多提出的方法还显示出关于区域,非对映和对映选择性转化的额外选择性,其中一个例子甚至通过动态动力学拆分进行。在此说明中提出的许多进步是由详细的机械研究(包括DFT计算,ESI-MS和原位IR实验)推动的,并使得我们的化学方法可用于本文所示的几个实例证明的目标导向的合成。一般来说,在合成手性化合物时,该研究主题已在过去几年中逐渐成熟,成为一种可行的选择,
更新日期:2016-07-25
中文翻译:
分支:铑催化的炔烃和丙二烯的烯丙基化
我们提出了一种新的有效策略,通过我们的工作组开发的一种通过Rh催化的异构化/加成反应,将炔烃和丙二烯进行原子经济转化为烯丙基官能化结构的方法。因此,我们的方法学使人们能够利用现代有机化学中有价值的重要结构类别,因为它具有进一步实现功能化的多功能性以及通过构建新的立体异构中心进行不对称合成的潜力。这种新方法是受到1980年代后期Werner进行的机械研究的启发,并基于Yamamoto和Trost的初步工作,为其他已建立的烯丙基功能化方法(如烯丙基取代或烯丙基氧化)提供了有吸引力的替代方法。与烯丙基氧化或取代相比,我们方法的主要优势在于固有的原子经济性,这既会产生化学计量的废物,而且在发生取代反应的情况下,还需要对原料进行预功能化。从发现使用Rh(I)/ DPEphos络合物作为催化剂体系发现羧酸与末端炔烃的高度支化-选择性偶联反应开始,在过去的5年中,我们能够不断扩展这种化学反应,引入各种(亲)亲核试剂通过选择合适的铑/双齿膦催化剂对炔烃和双键异构艾伦进行选择性C–O,C–S,C–N和C–C功能化。因此,有价值的化合物,例如支链烯丙基醚,砜,胺或γ,δ-不饱和酮已成功地以高收率和广泛的底物范围合成。除了铑固有的支化选择性外,许多提出的方法还显示出关于区域,非对映和对映选择性转化的额外选择性,其中一个例子甚至通过动态动力学拆分进行。在此说明中提出的许多进步是由详细的机械研究(包括DFT计算,ESI-MS和原位IR实验)推动的,并使得我们的化学方法可用于本文所示的几个实例证明的目标导向的合成。一般来说,在合成手性化合物时,该研究主题已在过去几年中逐渐成熟,成为一种可行的选择,



















































 京公网安备 11010802027423号
京公网安备 11010802027423号