当前位置:
X-MOL 学术
›
Int. J. Quantum Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
First‐principle study of the properties in BaTiO3 and the electronic structure of H2O adsorption on BaTiO3
International Journal of Quantum Chemistry ( IF 2.3 ) Pub Date : 2020-12-24 , DOI: 10.1002/qua.26576 Jiao Wang 1, 2 , Zhiguo Xing 2 , Zhenlin Lu 1 , Kaining Ding 3 , Haidou Wang 2
International Journal of Quantum Chemistry ( IF 2.3 ) Pub Date : 2020-12-24 , DOI: 10.1002/qua.26576 Jiao Wang 1, 2 , Zhiguo Xing 2 , Zhenlin Lu 1 , Kaining Ding 3 , Haidou Wang 2
Affiliation
|
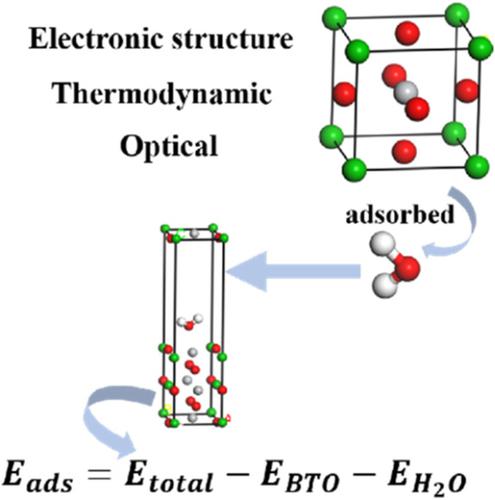
The properties of BaTiO3 and the electronic structure of H2O adsorption on BaTiO3 were investigated by using first principle calculations based on density functional theory. The calculated results show that the orbital hybridization between Ti and O is stronger than that between Ba and O. With the increase of energy, reflex points in the form of random fluctuation and the anisotropy of crystal clear. Enthalpy and heat capacity varies with temperature rise, while free energy fall. H bonds are formed between the hydrogen of water and the surface oxygen of BaTiO3 and between the hydrogen of hydroxyl and the surface oxygen of BaTiO3. The adsorption process of BaTiO3 adsorbing H2O is a physical process and ΔH < 0, ΔS < 0.
中文翻译:

BaTiO3性质和BaTiO3上H2O吸附电子结构的第一性原理研究
通过基于密度泛函理论的第一性原理计算,研究了BaTiO 3的性质和H 2 O在BaTiO 3上的吸附电子结构。计算结果表明,Ti和O之间的轨道杂化强于Ba和O之间的杂化。随着能量的增加,反射点以随机波动的形式出现,并具有透明的各向异性。焓和热容随温度升高而变化,而自由能下降。H键被水的氢气和钛酸钡的表面氧之间形成3和羟基的氢和钛酸钡的表面氧之间3。BaTiO 3吸附H的吸附过程2 O是一个物理过程,ΔH <0,ΔS <0。
更新日期:2020-12-24
中文翻译:
BaTiO3性质和BaTiO3上H2O吸附电子结构的第一性原理研究
通过基于密度泛函理论的第一性原理计算,研究了BaTiO 3的性质和H 2 O在BaTiO 3上的吸附电子结构。计算结果表明,Ti和O之间的轨道杂化强于Ba和O之间的杂化。随着能量的增加,反射点以随机波动的形式出现,并具有透明的各向异性。焓和热容随温度升高而变化,而自由能下降。H键被水的氢气和钛酸钡的表面氧之间形成3和羟基的氢和钛酸钡的表面氧之间3。BaTiO 3吸附H的吸附过程2 O是一个物理过程,ΔH <0,ΔS <0。




















































 京公网安备 11010802027423号
京公网安备 11010802027423号