当前位置:
X-MOL 学术
›
Inorg. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Transitioning from Intraligand π,π* to Charge-Transfer Excited States Using Thiophene-Based Donor–Acceptor Systems
Inorganic Chemistry ( IF 4.3 ) Pub Date : 2020-12-21 , DOI: 10.1021/acs.inorgchem.0c02555 James R. W. McLay 1 , Joshua J. Sutton 1 , Georgina E. Shillito 1 , Christopher B. Larsen 1 , Gregory S. Huff 1 , Nigel T. Lucas 1 , Keith C. Gordon 1
Inorganic Chemistry ( IF 4.3 ) Pub Date : 2020-12-21 , DOI: 10.1021/acs.inorgchem.0c02555 James R. W. McLay 1 , Joshua J. Sutton 1 , Georgina E. Shillito 1 , Christopher B. Larsen 1 , Gregory S. Huff 1 , Nigel T. Lucas 1 , Keith C. Gordon 1
Affiliation
|
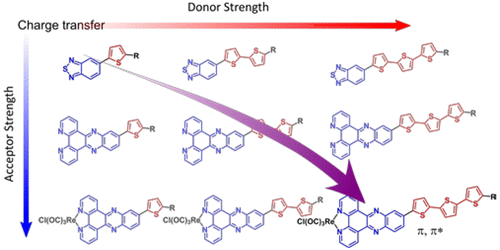
A series of electron donor–acceptor compounds are reported in which both the donor and acceptor strengths are systematically altered using mono-, bi-, and terthiophene as donors and benzo[c][1,2,5]thiadiazole (btd), dipyrido[3,2-a:2′,3′-c]phenazine (dppz), and the corresponding rhenium(I) complex, [ReCl(CO)3(dppz)], as acceptors. The electronic properties of the compounds are characterized using electrochemistry, electronic absorbance and emission spectroscopies, and transient absorption spectroscopy. The effect of donor and acceptor strengths on frontier molecular orbital localization and on the charge-transfer (CT) character of optical transitions is modeled using density functional theory (DFT) calculations. The electronic absorption spectra of the compounds investigated are dominated by intraligand charge-transfer (ILCT) transitions, where the CT character is shown to increase across the series from mono- to bi- to terthiophene but not significantly across the acceptor series. Emission is shown to originate from the absorbing state. Long-lived nonemissive states have been observed using transient absorption spectroscopy and assigned using triplet-state DFT calculations, which indicate that the lowest energy excited state has more thiophene-localized π,π* character with an increasing number of appended thiophenes.
中文翻译:

使用基于噻吩的供体-受体系统从配位体π,π*过渡到电荷转移激发态
报道了一系列电子供体-受体化合物,其中使用单噻吩,双噻吩和对噻吩作为供体和苯并[ c ] [1,2,5]噻二唑(btd),双嘧啶系统地改变了供体和受体的强度。[3,2- a:2',3'- c ]吩嗪(dppz)和相应的rh(I)配合物[ReCl(CO)3(dppz)],作为接受者。使用电化学,电子吸收光谱和发射光谱以及瞬态吸收光谱对化合物的电子性质进行表征。使用密度泛函理论(DFT)计算来建模供体和受体强度对前沿分子轨道定位和光学跃迁的电荷转移(CT)特性的影响。所研究化合物的电子吸收光谱主要由配体内电荷转移(ILCT)跃迁控制,其中CT特性在从单噻吩到双噻吩到三噻吩的整个过程中均显示出增加,但在受体系列中却没有显着增加。已显示出发射源于吸收状态。
更新日期:2021-01-04
中文翻译:
使用基于噻吩的供体-受体系统从配位体π,π*过渡到电荷转移激发态
报道了一系列电子供体-受体化合物,其中使用单噻吩,双噻吩和对噻吩作为供体和苯并[ c ] [1,2,5]噻二唑(btd),双嘧啶系统地改变了供体和受体的强度。[3,2- a:2',3'- c ]吩嗪(dppz)和相应的rh(I)配合物[ReCl(CO)3(dppz)],作为接受者。使用电化学,电子吸收光谱和发射光谱以及瞬态吸收光谱对化合物的电子性质进行表征。使用密度泛函理论(DFT)计算来建模供体和受体强度对前沿分子轨道定位和光学跃迁的电荷转移(CT)特性的影响。所研究化合物的电子吸收光谱主要由配体内电荷转移(ILCT)跃迁控制,其中CT特性在从单噻吩到双噻吩到三噻吩的整个过程中均显示出增加,但在受体系列中却没有显着增加。已显示出发射源于吸收状态。



















































 京公网安备 11010802027423号
京公网安备 11010802027423号