Journal of Molecular Liquids ( IF 5.3 ) Pub Date : 2020-12-17 , DOI: 10.1016/j.molliq.2020.115092 Antonio García Martínez , Pedro C. Gómez , Santiago de la Moya , Hans-Ullrich Siehl
|
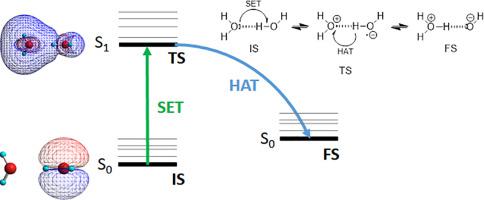
The rate constants for the autoprotolysis (kA) and corresponding ions recombination (kR) reaction of water and related protic liquids are computed considering both processes as reversible, sequential two-step reactions according to a breaking mechanism, which is supported both experimentally and computationally. The first step, according to the Marcus Theory (MT), consists of an horizontal single electron transfer (SET) occurring in the most stable water dimer, the reaction initial state (IS), to produce the transition state (TS). The second step is a hydrogen atom transfer (HAT), taking place consecutively at the said TS, leading to the reaction final state (FS) after relaxation. The optimized structure of the endergonic FS was calculated by means of the Time Dependent Density Functional Theory (TD-DFT). The determination of the free energy of activation for the autoprotolysis and for the corresponding ions recombination (∆GA≠ and ∆GR≠, respectively) was carried out following the outer-sphere adiabatic MT. The generality of both the proposed mechanism and used computational method is supported by the accurate calculation (MAPE ca. 13%) of the autoprotolysis constants (pKA) of water and five protic liquids (small alcohols and ammonia), covering a range of ca. 15 pKA units. Alternative feasible water mechanisms such as the Single Proton Transfer (SPT) mechanism, based on the non-concerted proton transfer between two water molecules, and the Concerted SET and Hydrogen Atom Transfer (CSETHAT) mechanism were also considered. Nonetheless, all these alternative mechanisms were discarded due to their computed slower autoprotolysis kinetics.
中文翻译:
基于Marcus理论和TD-DFT方法揭示水自质分解的机理
自分解(k A)和相应离子重组(k R)的速率常数。水和相关质子液体的反应是根据断裂机理将两个过程都视为可逆的,连续的两步反应来计算的,这在实验和计算上都得到了支持。根据马库斯理论(MT),第一步包括在最稳定的水二聚体中发生的水平单电子转移(SET),即反应初始状态(IS),以生成过渡态(TS)。第二步是在所述TS处连续发生氢原子转移(HAT),导致松弛后反应最终状态(FS)。通过时间相关密度泛函理论(TD-DFT)计算了endergonic FS的优化结构。自动质子化和相应离子重组的活化自由能的确定(∆G A ≠和∆ G R ≠)分别在外球面绝热MT之后进行。所提出的机制和使用的计算方法两者的通用性是通过精确计算(MAPE支持约的质子自迁移反应常数(PK的13%)甲的水)和五个质子液体(小醇和氨),覆盖的范围内的CA 。15的pK一单位。还考虑了其他可行的水机制,例如基于两个水分子之间未经证实的质子转移的单质子转移(SPT)机制,以及协同SET和氢原子转移(CSETHAT)机制。尽管如此,所有这些替代机制由于其计算的较慢的自体分解动力学而被丢弃。





























 京公网安备 11010802027423号
京公网安备 11010802027423号