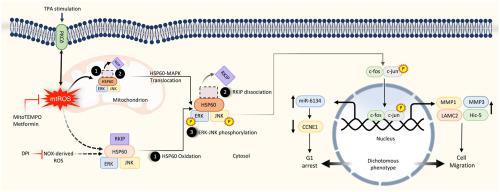
蛋白激酶 C (PKC) 和活性氧 (ROS) 都是众所周知的信号信使,它们相互交叉激活丝裂原活化蛋白激酶 (MAPKs) 以促进肝细胞癌 (HCC) 的进展。然而,潜在的机制尚未得到很好的阐明。特别是,是否涉及线粒体 ROS (mtROS) 以及它如何触发 MAPK 信号是很有趣的。在这项研究中,我们发现 mtROS 的产生和 MAPK 的磷酸化是由 PKCδ 在用肿瘤启动子 12- O治疗的 HCC 中介导的。-十四烷酰佛波醇 13-乙酸酯 (TPA)。热休克蛋白 60 (HSP60) 是线粒体中的一种伴侣蛋白,是 TPA 处理的 HCC 中氧化的主要蛋白质。此外,HSP60 的消耗或 HSP60 半胱氨酸突变体的表达阻止了 TPA 诱导的 MAPK 磷酸化。为了描述 HSP60 如何介导 MAPK 活化,研究了 Raf 激酶抑制蛋白 (RKIP) 的作用,Raf 激酶抑制蛋白 (RKIP) 是 MAPK 的负调节剂。TPA 在线粒体和胞质溶胶中将 RKIP 从 HSP60 解离,同时 HSP60 和 MAPK 从线粒体易位到胞质溶胶,这与胞质溶胶中 MAPK 的强磷酸化有关。此外,TPA 诱导了相反的 HCC 表型变化、G1 细胞周期停滞和细胞迁移,而 mtROS 清除剂和 PKCδ 和 HSP60 的消耗阻止了这些变化。始终如一,2、通过PKCδ/mtROS/HSP60/MAPK轴抑制细胞周期调节因子cyclin E1 (CCNE1)。最后,TPA 诱导的迁移相关基因表达需要 c-jun 和 c-fos,而一种新的 microRNA,miR-6134,负责 TPA 诱导的 CCNE1 抑制。总之,PKCδ 与 mtROS 相互作用触发 HSP60 氧化以释放 RKIP 以激活 MAPK,调节基因表达以进行迁移和 HCC 中的 G1 细胞周期停滞。针对 PKCδ、RKIP 和 HSP60 等关键参与者的靶向治疗有望预防 HCC 进展。
 "点击查看英文标题和摘要"
"点击查看英文标题和摘要"
PKCδ mediates mitochondrial ROS generation and oxidation of HSP60 to relieve RKIP inhibition on MAPK pathway for HCC progression
Both protein kinase C (PKC) and reactive oxygen species (ROS) are well-known signaling messengers cross-talking with each other to activate mitogen-activated protein kinases (MAPKs) for progression of hepatocellular carcinoma (HCC). However, the underlying mechanisms are not well elucidated. Especially, whether mitochondrial ROS (mtROS) is involved and how it triggers MAPK signaling are intriguing. In this study, we found mtROS generation and phosphorylation of MAPKs were mediated by PKCδ in HCCs treated with the tumor promoter 12-O-tetradecanoyl-phorbol-13-acetate (TPA). Heat shock protein 60 (HSP60), one of the chaperones in mitochondria was the major protein oxidized in TPA-treated HCCs. Moreover, depletion of HSP60 or expression of HSP60 cysteine mutant prevented TPA-induced phosphorylation of MAPKs. To delineate how HSP60 mediated MAPK activation, the role of Raf kinase inhibitor protein (RKIP), a negative regulator of MAPK, was investigated. TPA dissociated RKIP from HSP60 in both mitochondria and cytosol, concurrently with translocation of HSP60 and MAPK from mitochondria to cytosol, which was associated with robust phosphorylation of MAPKs in the cytosol. Moreover, TPA induced opposite phenotypical changes of HCCs, G1 cell cycle arrest, and cell migration, which were prevented by mtROS scavengers and depletion of PKCδ and HSP60. Consistently, TPA increased the migration-related genes, hydrogen peroxide inducible clone5, matrix metalloproteinase-1/3, laminin2, and suppressed the cell cycle regulator cyclin E1 (CCNE1) via PKCδ/mtROS/HSP60/MAPK-axis. Finally, c-jun and c-fos were required for TPA-induced expression of the migration-related genes and a novel microRNA, miR-6134, was responsible for TPA-induced suppression of CCNE1. In conclusion, PKCδ cross-talked with mtROS to trigger HSP60 oxidation for release of RKIP to activate MAPK, regulating gene expression for migration, and G1 cell cycle arrest in HCC. Targeted therapy aiming at key players like PKCδ, RKIP, and HSP60 is promising for preventing HCC progression.


































 京公网安备 11010802027423号
京公网安备 11010802027423号