当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Deciphering Reaction Determinants of Altered-Activity CYP2D6 Variants by Well-Tempered Metadynamics Simulation and QM/MM Calculations
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-12-03 , DOI: 10.1021/acs.jcim.0c01091
Charleen G Don 1 , Martin Smieško 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-12-03 , DOI: 10.1021/acs.jcim.0c01091
Charleen G Don 1 , Martin Smieško 1
Affiliation
|
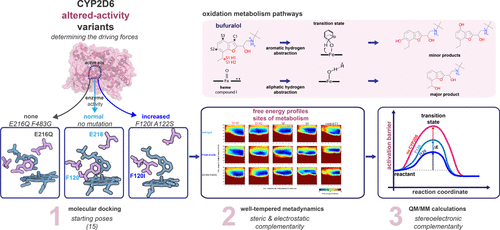
The xenobiotic metabolizing enzyme CYP2D6 is the P450 cytochrome family member with the highest rate of polymorphism. This causes changes in the enzyme activity and specificity, which can ultimately lead to adverse reactions during drug treatment. To avoid or lower CYP-related toxicity risks, prediction of the most likely positions within a molecule where a metabolic reaction might occur is paramount. In order to obtain accurate predictions, it is crucial to understand all phenomena within the active site of the enzyme that contribute to an efficient substrate recognition and the subsequent catalytic reaction together with their relative weight within the overall thermodynamic context. This study aims to define the weight of the driving forces upon the C–H bond activation within CYP2D6 wild-type and a clinically relevant allelic variant with increased activity (CYP2D6*53) featuring two amino acid mutations in close vicinity of the heme. First, we investigated the steric and electrostatic complementarity of the substrate bufuralol using well-tempered metadynamics simulations with the aim to obtain the free energy profiles for each site of metabolism (SoM) within the different active sites. Second, the stereoelectronic complementarity was determined for each SoM within the two different active-site environments. Relying on the well-tempered metadynamics simulation energy profiles of each SoM, we identified the binding mode that was closest to the preferred transition-state geometry for efficient C–H bond activation. The binding modes were then used as starting structures for the quantum mechanics/molecular mechanics calculations performed to quantify the corresponding activation barriers. Our results show the relevance of the steric component in orienting the SoM in an energetically accessible position toward the heme. However, the corresponding intrinsic reactivity and electronic complementarity within the active site must be accurately evaluated in order to obtain a meaningful reaction prediction, from which the predominant SoM can be determined. The F120I mutation lowered the activation barrier for the major site and one of the minor SoMs. However, it had an impact neither on the CYP2D6 enantioselectivity preference of the oxidation reaction nor on the stereoselectivity from the substrate point of view.
中文翻译:

脾气暴躁的动力学模拟和QM / MM计算方法对活性改变的CYP2D6变体的破译反应行列式
异种代谢酶CYP2D6是P450细胞色素家族成员,具有最高的多态性。这导致酶活性和特异性的改变,最终可能导致药物治疗期间的不良反应。为了避免或降低CYP相关的毒性风险,最重要的是预测分子中可能发生代谢反应的最可能位置。为了获得准确的预测,至关重要的是要了解酶活性位点中有助于有效底物识别和后续催化反应的所有现象,以及它们在整个热力学范围内的相对重量。CYP2D6 * 53)在血红素附近存在两个氨基酸突变。首先,我们使用经过精心调配的元动力学模拟研究了底富卢草醇的空间和静电互补性,目的是获得不同活性位点内每个代谢位点(SoM)的自由能分布。其次,在两个不同的活动站点环境中为每个SoM确定立体声电子互补性。依靠每个SoM的良好的动力学模拟能量分布,我们确定了最有效的C–H键活化最接近首选过渡态几何结构的结合模式。然后将结合模式用作进行量子力学/分子力学计算以量化相应的激活势垒的起始结构。我们的结果表明,在将SoM定位在朝向血红素的能量可及位置时,空间成分的相关性。但是,必须准确评估活性位点内的相应内在反应性和电子互补性,以获得有意义的反应预测,从而可以确定主要的SoM。F120I突变降低了主要位点和一个次要SoM的激活屏障。然而,从底物的观点来看,它既不影响氧化反应的CYP2D6对映选择性,也不影响立体选择性。必须准确评估活性位点内相应的固有反应性和电子互补性,以获得有意义的反应预测,从而可以确定主要的SoM。F120I突变降低了主要位点和一个次要SoM的激活屏障。然而,从底物的观点来看,它既不影响氧化反应的CYP2D6对映选择性,也不影响立体选择性。必须准确评估活性位点内相应的固有反应性和电子互补性,以获得有意义的反应预测,从而可以确定主要的SoM。F120I突变降低了主要位点和一个次要SoM的激活屏障。然而,从底物的观点来看,它既不影响氧化反应的CYP2D6对映选择性,也不影响立体选择性。
更新日期:2020-12-28
中文翻译:
脾气暴躁的动力学模拟和QM / MM计算方法对活性改变的CYP2D6变体的破译反应行列式
异种代谢酶CYP2D6是P450细胞色素家族成员,具有最高的多态性。这导致酶活性和特异性的改变,最终可能导致药物治疗期间的不良反应。为了避免或降低CYP相关的毒性风险,最重要的是预测分子中可能发生代谢反应的最可能位置。为了获得准确的预测,至关重要的是要了解酶活性位点中有助于有效底物识别和后续催化反应的所有现象,以及它们在整个热力学范围内的相对重量。CYP2D6 * 53)在血红素附近存在两个氨基酸突变。首先,我们使用经过精心调配的元动力学模拟研究了底富卢草醇的空间和静电互补性,目的是获得不同活性位点内每个代谢位点(SoM)的自由能分布。其次,在两个不同的活动站点环境中为每个SoM确定立体声电子互补性。依靠每个SoM的良好的动力学模拟能量分布,我们确定了最有效的C–H键活化最接近首选过渡态几何结构的结合模式。然后将结合模式用作进行量子力学/分子力学计算以量化相应的激活势垒的起始结构。我们的结果表明,在将SoM定位在朝向血红素的能量可及位置时,空间成分的相关性。但是,必须准确评估活性位点内的相应内在反应性和电子互补性,以获得有意义的反应预测,从而可以确定主要的SoM。F120I突变降低了主要位点和一个次要SoM的激活屏障。然而,从底物的观点来看,它既不影响氧化反应的CYP2D6对映选择性,也不影响立体选择性。必须准确评估活性位点内相应的固有反应性和电子互补性,以获得有意义的反应预测,从而可以确定主要的SoM。F120I突变降低了主要位点和一个次要SoM的激活屏障。然而,从底物的观点来看,它既不影响氧化反应的CYP2D6对映选择性,也不影响立体选择性。必须准确评估活性位点内相应的固有反应性和电子互补性,以获得有意义的反应预测,从而可以确定主要的SoM。F120I突变降低了主要位点和一个次要SoM的激活屏障。然而,从底物的观点来看,它既不影响氧化反应的CYP2D6对映选择性,也不影响立体选择性。
































 京公网安备 11010802027423号
京公网安备 11010802027423号