当前位置:
X-MOL 学术
›
J. Mol. Struct.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
寻求更好地理解乙醇与镁和锌卟啉的配位:结合实验和理论研究
Journal of Molecular Structure ( IF 4.0 ) Pub Date : 2021-04-01 , DOI: 10.1016/j.molstruc.2020.129646
Bishnu Prasad Borah , Smita Majumder , Karishma Devi Borah , Jagannath Bhuyan
Journal of Molecular Structure ( IF 4.0 ) Pub Date : 2021-04-01 , DOI: 10.1016/j.molstruc.2020.129646
Bishnu Prasad Borah , Smita Majumder , Karishma Devi Borah , Jagannath Bhuyan
|
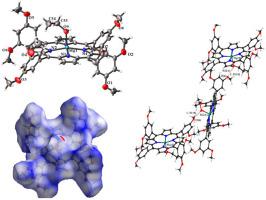
摘要 合成了一种乙醇配位镁卟啉[Mg(TDMPP)(C2H5OH)], (TDMPP = 5,10,15,20-四(3,5-二甲氧基苯基)卟啉), 1 并通过单晶X射线衍射表征方法以及其他标准光谱技术。这是四苯基卟啉衍生物的醇结合镁卟啉的第一个已知晶体结构。该化合物的晶体结构显示轴向乙醇分子的 OH 基团与相邻卟啉分子的外围甲氧基之间存在弱氢键相互作用 (2.730(4) A)。这种氢键导致形成弱的一维超分子结构。还进行了荧光和电化学研究。探讨使用锌卟啉研究叶绿素型镁卟啉的弊端,使用 DFT 水平对化合物 [Mg(TDMPP)(C2H5OH)], 1 及其锌对应物 [Mg(TDMPP)(C2H5OH)], 2 的理论研究进行了比较。 理论计算包括几何优化、前沿分子能量轨道 (FMO)、电子跃迁和全球化学指数。从锌和镁卟啉的理论计算得出的全局反应性指数如 η、μ 和 ω 支持镁卟啉比锌类似物更具反应性。此外,Hirshfeld 表面分析用于量化化合物 1 晶格中存在的弱分子间相互作用。前沿分子轨道 (FMO)、电子跃迁和全球化学指数的能量。从锌和镁卟啉的理论计算得出的全局反应性指数如 η、μ 和 ω 支持镁卟啉比锌类似物更具反应性。此外,Hirshfeld 表面分析用于量化化合物 1 晶格中存在的弱分子间相互作用。前沿分子轨道 (FMO)、电子跃迁和全球化学指数的能量。从锌和镁卟啉的理论计算得出的全局反应性指数如 η、μ 和 ω 支持镁卟啉比锌类似物更具反应性。此外,Hirshfeld 表面分析用于量化化合物 1 晶格中存在的弱分子间相互作用。

"点击查看英文标题和摘要"
更新日期:2021-04-01
"点击查看英文标题和摘要"
































 京公网安备 11010802027423号
京公网安备 11010802027423号