当前位置:
X-MOL 学术
›
Comp. Mater. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Theoretical investigation of spin-dependent electron transport properties of dibromobenzene based positional isomers
Computational Materials Science ( IF 3.1 ) Pub Date : 2021-02-01 , DOI: 10.1016/j.commatsci.2020.110109 A. Aadhityan , C. Preferencial Kala , D. John Thiruvadigal
Computational Materials Science ( IF 3.1 ) Pub Date : 2021-02-01 , DOI: 10.1016/j.commatsci.2020.110109 A. Aadhityan , C. Preferencial Kala , D. John Thiruvadigal
|
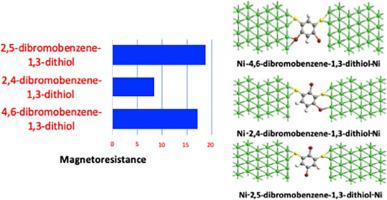
Abstract Understanding the spin-dependent electron transport through a single molecular junction will provide in-depth knowledge to construct efficient molecular spintronic devices. The electron transport in such junction highly depends upon the structure of the molecule. The Density Functional Theory (DFT) combined with Non-Equilibrium Green's Function (NEGF) utilized to investigate the spin-dependent electron transport properties of positional isomers namely 4,6‐dibromobenzene‐1,3‐dithiol; 2,4‐dibromobenzene‐1,3‐dithiol and 2,5-dibromobenzene-1,3-dithiol. We have calculated several parameters such as the density of states, current–voltage characteristic, magnetoresistance effect etc. of these molecular junctions to understand the nature of electron transport. The projected density of states of the molecule and the total density of states shows that these molecules have good coupling with the electrode. Under the applied bias, we observed a variation of spin up and spin down transmission which leads to magnetoresistance effect in these molecular junctions. Among these molecules, the 2,5-dibromobenzene-1,3-thiol molecular junction shows a higher magnetoresistance effect than 4,6‐dibromobenzene‐1,3‐dithiol and 2,4‐dibromobenzene‐1,3‐dithiol. Hence, 2,5-dibromobenzene-1,3-thiol can be a good candidate for spintronic applications.
中文翻译:

基于二溴苯的位置异构体的自旋相关电子传输特性的理论研究
摘要 了解通过单个分子结的自旋相关电子传输将为构建高效的分子自旋电子器件提供深入的知识。这种结中的电子传输高度依赖于分子的结构。密度泛函理论 (DFT) 与非平衡格林函数 (NEGF) 相结合,用于研究位置异构体即 4,6-二溴苯-1,3-二硫醇的自旋相关电子传输特性;2,4-二溴苯-1,3-二硫醇和2,5-二溴苯-1,3-二硫醇。我们计算了几个参数,例如这些分子结的态密度、电流-电压特性、磁阻效应等,以了解电子传输的性质。分子的投影态密度和总态密度表明这些分子与电极具有良好的耦合。在施加的偏压下,我们观察到自旋向上和自旋向下传输的变化,这导致这些分子结中的磁阻效应。在这些分子中,2,5-二溴苯-1,3-硫醇分子结显示出比4,6-二溴苯-1,3-二硫醇和2,4-二溴苯-1,3-二硫醇更高的磁阻效应。因此,2,5-二溴苯-1,3-硫醇可以成为自旋电子应用的良好候选者。3-硫醇分子结显示出比4,6-二溴苯-1,3-二硫醇和2,4-二溴苯-1,3-二硫醇更高的磁阻效应。因此,2,5-二溴苯-1,3-硫醇可以成为自旋电子应用的良好候选者。3-硫醇分子结显示出比4,6-二溴苯-1,3-二硫醇和2,4-二溴苯-1,3-二硫醇更高的磁阻效应。因此,2,5-二溴苯-1,3-硫醇可以成为自旋电子应用的良好候选者。
更新日期:2021-02-01
中文翻译:
基于二溴苯的位置异构体的自旋相关电子传输特性的理论研究
摘要 了解通过单个分子结的自旋相关电子传输将为构建高效的分子自旋电子器件提供深入的知识。这种结中的电子传输高度依赖于分子的结构。密度泛函理论 (DFT) 与非平衡格林函数 (NEGF) 相结合,用于研究位置异构体即 4,6-二溴苯-1,3-二硫醇的自旋相关电子传输特性;2,4-二溴苯-1,3-二硫醇和2,5-二溴苯-1,3-二硫醇。我们计算了几个参数,例如这些分子结的态密度、电流-电压特性、磁阻效应等,以了解电子传输的性质。分子的投影态密度和总态密度表明这些分子与电极具有良好的耦合。在施加的偏压下,我们观察到自旋向上和自旋向下传输的变化,这导致这些分子结中的磁阻效应。在这些分子中,2,5-二溴苯-1,3-硫醇分子结显示出比4,6-二溴苯-1,3-二硫醇和2,4-二溴苯-1,3-二硫醇更高的磁阻效应。因此,2,5-二溴苯-1,3-硫醇可以成为自旋电子应用的良好候选者。3-硫醇分子结显示出比4,6-二溴苯-1,3-二硫醇和2,4-二溴苯-1,3-二硫醇更高的磁阻效应。因此,2,5-二溴苯-1,3-硫醇可以成为自旋电子应用的良好候选者。3-硫醇分子结显示出比4,6-二溴苯-1,3-二硫醇和2,4-二溴苯-1,3-二硫醇更高的磁阻效应。因此,2,5-二溴苯-1,3-硫醇可以成为自旋电子应用的良好候选者。
































 京公网安备 11010802027423号
京公网安备 11010802027423号