当前位置:
X-MOL 学术
›
ACS Catal.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Mechanistic Insights into Hydroformylation Catalyzed by Cationic Cobalt(II) Complexes: In Silico Modification of the Catalyst System
ACS Catalysis ( IF 11.3 ) Pub Date : 2020-11-10 , DOI: 10.1021/acscatal.0c03161 Jiandong Guo 1, 2 , Dongju Zhang 2 , Xiaotai Wang 1, 3
ACS Catalysis ( IF 11.3 ) Pub Date : 2020-11-10 , DOI: 10.1021/acscatal.0c03161 Jiandong Guo 1, 2 , Dongju Zhang 2 , Xiaotai Wang 1, 3
Affiliation
|
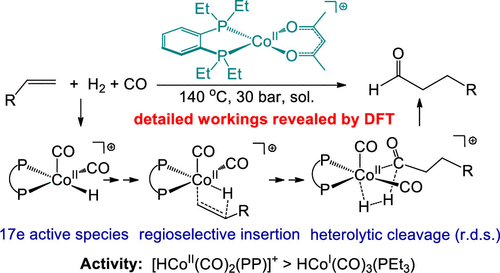
The hydroformylation reaction is used on a large industrial scale to convert olefins and synthesis gas (CO + H2) into aldehydes. Researchers have recently discovered that a class of cationic Co(II) complexes of the formula [CoII(PP)(acac)]+ (PP = diphosphine, acac = acetylacetonate) can catalyze hydroformylation with activity approaching that of the widely used rhodium catalysts (Hood, D. M. et al. Science 2020, 367, 542−548). This density functional theory (DFT) study reveals the detailed workings of the cationic Co(II) catalyst system. The precatalyst [CoII(PP)(acac)]+ is initiated by reacting with H2 and CO to generate active species [HCoII(CO)2(PP)]+. In comparison with the 18-electron neutral Co(I) catalytic species HCoI(CO)3(PR3), these cationic Co(II) species, with their unique 17-electron and square pyramidal structure, invoke a lower-energy pathway through different elementary steps such as associative alkene uptake and heterolytic H2 cleavage. The regioselectivity for linear aldehyde products is due to a combination of electronic and steric effects that favor the anti-Markovnikov insertion of a terminal alkene into the Co–H bond. DFT calculations predict that addition of PMe3 would facilitate the precatalyst initiation, thereby decreasing the reaction temperature or shortening the induction period. The insights gained by this theoretical study can be useful for the further development of hydroformylation catalysts.
中文翻译:

阳离子钴(II)配合物催化加氢甲酰化的机理研究:催化体系的计算机修饰
加氢甲酰化反应用于大规模工业中,以将烯烃和合成气(CO + H 2)转化为醛。研究人员最近发现,一类式为[Co II(PP)(acac)] +(PP =二膦,acac =乙酰丙酮酸)的阳离子Co(II)配合物可以催化加氢甲酰化,其活性接近于广泛使用的铑催化剂。 (罩,DM等人科学 2020,367,542-548)。该密度泛函理论(DFT)研究揭示了阳离子Co(II)催化剂体系的详细工作原理。通过与H 2反应引发前催化剂[Co II(PP)(acac)] +和CO生成活性物质[HCo II(CO)2(PP)] +。与18电子中性Co(I)催化物质HCo I(CO)3(PR 3)相比,这些阳离子Co(II)物质具有独特的17电子和方形金字塔结构,调用了较低的能量途径通过不同的基本步骤,例如缔合烯烃吸收和H 2裂解。线性醛产物的区域选择性是由于电子和空间效应的结合,有利于将末端烯烃的反马尔科夫尼可夫插入Co-H键中。DFT计算预测添加PMe 3这将促进预催化剂的引发,从而降低反应温度或缩短诱导时间。通过该理论研究获得的见识可用于进一步开发加氢甲酰化催化剂。
更新日期:2020-11-21
中文翻译:
阳离子钴(II)配合物催化加氢甲酰化的机理研究:催化体系的计算机修饰
加氢甲酰化反应用于大规模工业中,以将烯烃和合成气(CO + H 2)转化为醛。研究人员最近发现,一类式为[Co II(PP)(acac)] +(PP =二膦,acac =乙酰丙酮酸)的阳离子Co(II)配合物可以催化加氢甲酰化,其活性接近于广泛使用的铑催化剂。 (罩,DM等人科学 2020,367,542-548)。该密度泛函理论(DFT)研究揭示了阳离子Co(II)催化剂体系的详细工作原理。通过与H 2反应引发前催化剂[Co II(PP)(acac)] +和CO生成活性物质[HCo II(CO)2(PP)] +。与18电子中性Co(I)催化物质HCo I(CO)3(PR 3)相比,这些阳离子Co(II)物质具有独特的17电子和方形金字塔结构,调用了较低的能量途径通过不同的基本步骤,例如缔合烯烃吸收和H 2裂解。线性醛产物的区域选择性是由于电子和空间效应的结合,有利于将末端烯烃的反马尔科夫尼可夫插入Co-H键中。DFT计算预测添加PMe 3这将促进预催化剂的引发,从而降低反应温度或缩短诱导时间。通过该理论研究获得的见识可用于进一步开发加氢甲酰化催化剂。






























 京公网安备 11010802027423号
京公网安备 11010802027423号