当前位置:
X-MOL 学术
›
Inorg. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
31P Chemical Shifts in Ru(II) Phosphine Complexes. A Computational Study of the Influence of the Coordination Sphere
Inorganic Chemistry ( IF 4.3 ) Pub Date : 2020-11-06 , DOI: 10.1021/acs.inorgchem.0c02256 Christophe Raynaud 1 , Eliott Norbert-Agaisse 1 , Brian R. James 2 , Odile Eisenstein 1, 3
Inorganic Chemistry ( IF 4.3 ) Pub Date : 2020-11-06 , DOI: 10.1021/acs.inorgchem.0c02256 Christophe Raynaud 1 , Eliott Norbert-Agaisse 1 , Brian R. James 2 , Odile Eisenstein 1, 3
Affiliation
|
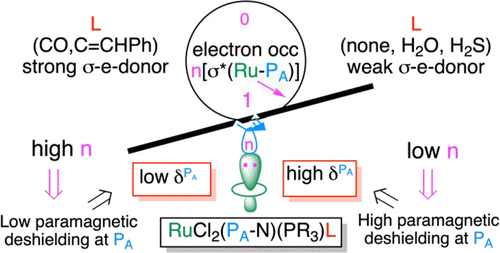
The NMR chemical shift has been the most versatile marker of chemical structures, by reflecting global and local electronic structures, and is very sensitive to any change within the chemical species. In this work, Ru(II) complexes with the same five ligands and a variable sixth ligand L (none, H2O, H2S, CH3SH, H2, N2, N2O, NO+, C═CHPh, and CO) are studied by using as the NMR reporter the phosphorus PA of a coordinated bidentate PA-N ligand (PA-N = o-diphenylphosphino-N,N′-dimethylaniline). The chemical shift of PA in RuCl2(PA-N)(PR3)(L) (R = phenyl, p-tolyl, or p-FC6H4) was shown to increase as the Ru–PA bond distance decreases, an observation that was not rationalized. This work, using density functional theory (DFT) calculations, reproduces reasonably well the observed 31P chemical shifts for these complexes and the correlation between the shifts and the Ru–PA bond distance as L varies. An interpretation of this correlation is proposed by using a natural chemical shift (NCS) analysis based on the natural bonding orbital (NBO) method. This analysis of the principal components of the chemical shift tensors shows how the σ-donating properties of L have a particularly high influence on the phosphine chemical shifts.
中文翻译:

Ru(II)膦配合物的31 P化学位移。协调域影响的计算研究
通过反映全局和局部电子结构,NMR化学位移已成为化学结构最通用的标记,并且对化学物种内的任何变化都非常敏感。在这项工作中,Ru(II)与相同的五个配体和一个可变的第六配体L(无,H 2 O,H 2 S,CH 3 SH,H 2,N 2,N 2 O,NO +,C═ CHPh配合,和CO),通过使用作为NMR记者的磷P研究甲的二齿配位P甲-N配体(P甲-N = Õ -diphenylphosphino- ñ,ñ′-二甲基苯胺)。结果表明,RuRu 2(P A -N)(PR 3)(L)(R =苯基,对甲苯基或p -FC 6 H 4)中P A的化学位移随Ru-P A键的增加而增加。距离减少,这是没有合理化的观察。这项工作使用密度泛函理论(DFT)计算,可以很好地再现观察到的这些复合物的31 P化学位移以及这些位移与Ru–P A之间的相关性。键距随着L的变化而变化。通过使用基于自然键轨道(NBO)方法的自然化学位移(NCS)分析,提出了对此相关性的解释。对化学位移张量的主要成分的分析表明,L的σ供电特性如何对膦的化学位移产生特别高的影响。
更新日期:2020-12-07
中文翻译:
Ru(II)膦配合物的31 P化学位移。协调域影响的计算研究
通过反映全局和局部电子结构,NMR化学位移已成为化学结构最通用的标记,并且对化学物种内的任何变化都非常敏感。在这项工作中,Ru(II)与相同的五个配体和一个可变的第六配体L(无,H 2 O,H 2 S,CH 3 SH,H 2,N 2,N 2 O,NO +,C═ CHPh配合,和CO),通过使用作为NMR记者的磷P研究甲的二齿配位P甲-N配体(P甲-N = Õ -diphenylphosphino- ñ,ñ′-二甲基苯胺)。结果表明,RuRu 2(P A -N)(PR 3)(L)(R =苯基,对甲苯基或p -FC 6 H 4)中P A的化学位移随Ru-P A键的增加而增加。距离减少,这是没有合理化的观察。这项工作使用密度泛函理论(DFT)计算,可以很好地再现观察到的这些复合物的31 P化学位移以及这些位移与Ru–P A之间的相关性。键距随着L的变化而变化。通过使用基于自然键轨道(NBO)方法的自然化学位移(NCS)分析,提出了对此相关性的解释。对化学位移张量的主要成分的分析表明,L的σ供电特性如何对膦的化学位移产生特别高的影响。






























 京公网安备 11010802027423号
京公网安备 11010802027423号