Free Radical Biology and Medicine ( IF 7.1 ) Pub Date : 2020-11-04 , DOI: 10.1016/j.freeradbiomed.2020.10.307 Li-Jing Tang 1 , Yuan-Jing Zhou 2 , Xiao-Ming Xiong 3 , Nian-Sheng Li 2 , Jie-Jie Zhang 4 , Xiu-Ju Luo 5 , Jun Peng 3
|
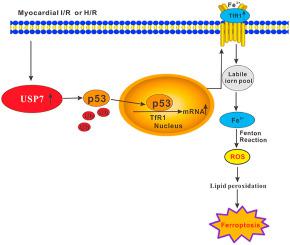
Iron overload triggers the ferroptosis in the heart following ischemia/reperfusion (I/R) and transferrin receptor 1 (TfR1) charges the cellular iron uptake. Bioinformatics analysis shows that the three molecules of ubiquitin-specific protease 7 (USP7), p53 and TfR1 form a unique pathway of USP7/p53/TfR1. This study aims to explore whether USP7/p53/TfR1 pathway promotes ferroptosis in rat hearts suffered I/R and the underlying mechanisms. The SD rat hearts were subjected to 1 h-ischemia plus 3 h-reperfusion, showing myocardial injury (increase in creatine kinase release, infarct size, myocardial fiber loss and disarray) and up-regulation of USP7, p53 and TfR1 concomitant with an increase of ferroptosis (reflecting by accumulation of iron and lipid peroxidation while decrease of glutathione peroxidase activity). Inhibition of USP7 activated p53 via suppressing deubiquitination, which led to down-regulation of TfR1, accompanied by the decreased ferroptosis and myocardial I/R injury. Next, H9c2 cells underwent hypoxia/reoxygenation (H/R) in vitro to mimic the myocardial I/R model in vivo. Consistent with the results in vivo, inhibition or knockdown of USP7 reduced the H/R injury (decrease of LDH release and necrosis) and enhanced the ubiquitination of p53 along with the decreased levels of p53 and TfR1 as well as the attenuated ferroptosis (manifesting as the decreased iron content and lipid peroxidation while the increased GPX activity). Knockdown of TfR1 inhibited H/R-induced ferroptosis without p53 deubiquitination. Based on these observations, we conclude that a novel pathway of USP7/p53/TfR1 has been identified in the I/R-treated rat hearts, where up-regulation of USP7promotes ferrptosis via activation of the p53/TfR1 pathway.
中文翻译:
泛素特异性蛋白酶7通过激活大鼠心脏缺血/再灌注后p53/TfR1通路促进铁死亡
铁超载会在缺血/再灌注 (I/R) 后触发心脏铁死亡,转铁蛋白受体 1 (TfR1) 对细胞铁的摄取产生充电。生物信息学分析表明,泛素特异性蛋白酶7(USP7)、p53和TfR1这三个分子形成了独特的USP7/p53/TfR1通路。本研究旨在探讨USP7/p53/TfR1通路是否促进I/R大鼠心脏铁死亡及其潜在机制。SD大鼠心脏经受1小时缺血加3小时再灌注,显示心肌损伤(肌酸激酶释放增加、梗死面积增加、心肌纤维丢失和混乱)和USP7、p53和TfR1的上调伴随增加铁死亡(反映铁和脂质过氧化的积累,同时谷胱甘肽过氧化物酶活性的降低)。抑制 USP7 通过抑制去泛素化激活 p53,导致 TfR1 的下调,伴随着铁死亡和心肌 I/R 损伤的减少。接下来,H9c2 细胞在体外进行缺氧/复氧 (H/R) 以模拟体内心肌 I/R 模型。与体内结果一致,USP7 的抑制或敲低减少了 H/R 损伤(减少 LDH 释放和坏死)并增强了 p53 的泛素化,同时降低了 p53 和 TfR1 的水平以及减弱的铁死亡(表现为铁含量和脂质过氧化降低,而 GPX 活性增加)。敲除 TfR1 可抑制 H/R 诱导的铁死亡而没有 p53 去泛素化。基于这些观察,我们得出结论,在 I/R 处理的大鼠心脏中发现了一种新的 USP7/p53/TfR1 通路,

















































 京公网安备 11010802027423号
京公网安备 11010802027423号