Computational and Theoretical Chemistry ( IF 3.0 ) Pub Date : 2020-11-01 , DOI: 10.1016/j.comptc.2020.113080 Xiajin Rao , Quanlong Si , Ting Shi , Xingbo Han , Shouxiao Ma
|
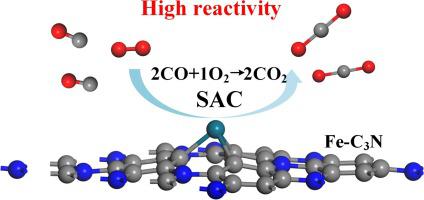
In this work, we investigate the CO oxidation on the Fe-doped N-vacancy C3N (Fe-C3N) monolayer by first-principle theory. Single Fe atom is doped on the N-vacancy site of C3N surface with a binding energy of −6.65 eV. The better performance of Fe-C3N upon O2 adsorption determines the preferred CO oxidation pathway by Eley-Rideal (ER) mechanism. Results indicate that the energy barriers in the two steps of ER mechanism are obtained as 0.45 and 0.47 eV, which could be feebly overcome at room temperature. Moreover, the physicochemical properties of the recovered Fe-C3N monolayer are unchanged compared with the original counterpart, manifesting that the CO oxidation on the Fe-C3N monolayer is energy-favorable at room temperature and the strong potential of Fe-C3N monolayer as a promising single-atom catalyst (SAC) for CO oxidation with high reactivity and low temperature. This work would be meaningful for the exploration of novel SAC in the field of heterogeneous catalysis.
中文翻译:
Fe掺杂的C 3 N单层作为低温和高反应活性的CO氧化的有希望的SAC
在这项工作中,我们通过第一原理研究了Fe掺杂N空位C 3 N(Fe-C 3 N)单层上的CO氧化。单个Fe原子以-6.65 eV的结合能掺杂在C 3 N表面的N空位上。Fe-C 3 N吸附O 2的性能更好,通过Eley-Rideal(ER)机理决定了优选的CO氧化途径。结果表明,在ER机理的两个步骤中获得的能垒分别为0.45和0.47eV,这在室温下可以克服。此外,回收的Fe-C 3的理化性质N单层与原始N单层相比没有变化,这表明Fe-C 3 N单层在室温下的CO氧化对能量有利,并且Fe-C 3 N单层作为有前途的单原子催化剂(SAC)具有强大的潜力)用于高活性和低温的CO氧化。这项工作对于探索多相催化领域的新型SAC具有重要意义。




















































 京公网安备 11010802027423号
京公网安备 11010802027423号