当前位置:
X-MOL 学术
›
J. Mol. Struct.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Synthesis, crystal structure, Hirshfeld surface, DFT calculations, Z-scan and nonlinear optical studies of novel flourinated hexahydropyrimidine
Journal of Molecular Structure ( IF 4.0 ) Pub Date : 2021-03-01 , DOI: 10.1016/j.molstruc.2020.129484 G. Kavitha , A. Dhandapani , B. Gunasekaran , M. Suresh
Journal of Molecular Structure ( IF 4.0 ) Pub Date : 2021-03-01 , DOI: 10.1016/j.molstruc.2020.129484 G. Kavitha , A. Dhandapani , B. Gunasekaran , M. Suresh
|
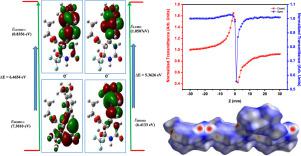
Abstract A new fluorinated hexahydropyrimidine was synthesized and its crystal structure was elucidated using single crystal X-ray diffraction technique. This compound, C15H17F3N2O6 crystallizes in monoclinic space group P21/c with cell parameters a = 10.6345(3) A, b = 14.7074(4) A, c = 10.6677(4) A, β= 101.453(2)° and V= 1635.27 (9) A3. In this compound the hexahydropyrimidine ring (C7/C8/C12/N1/C14/N2) adopts sofa conformation. The puckering parameters are Q = 0.5117A, Ɵ = 129.59° and φ =304.0802°. The analysis of Hirshfeld surface indicates the presence of hydrogen bonds C‒H•••F, C‒H•••O, O‒H•••O, N‒H•••O and π•••π stacking stabilizes the system. The energy optimized structure was calculated using Density Functional Theory (DFT) and were employed by hybrid functional theory (B3LYP) at 6-311++G(d,p) basis set in the Gaussian09_revision C0.1 program package. The natural bond orbital analysis results state that the highest energy transfer of 604.21 kJ/mol occur between the donor σ(O4‒H43) to acceptor σ*(C34‒H36) through carboxylate chain. Moreover, the vibrational assignments, global reactivity descriptors, energy gap and molecular electrostatic potential (MEP) also gives clear insight about chemical and biological activity of the molecule. The first hyperpolarizability value 7.2987 × 10−30 esu of the synthesized compound is twenty times greater than that of urea (0.3728 × 10−30 esu). The one electron excitation of the synthesized crystal calculated by the time dependent-density functional theory (TD-DFT) calculation and comparatively studied using recorded UV spectrum. The single beam Z-scan unit equipped with 532 nm continuous Nd:YAG wave laser with 5ns pulse width was used to measure third order nonlinear optical property reveal that the investigated molecule possess effective two photon absorption with higher effective absorption coefficient. The imaginary and real parts of the third-order susceptibility values determined as Im χ3 = 2.31 × 10−6 cm W−1 and Re χ3 = 8.74 × 10−8 cm2 W−1 respectively.
中文翻译:

新型氟化六氢嘧啶的合成、晶体结构、Hirshfeld 表面、DFT 计算、Z 扫描和非线性光学研究
摘要 合成了一种新的氟化六氢嘧啶,并利用单晶X射线衍射技术阐明了其晶体结构。该化合物 C15H17F3N2O6 在单斜空间群 P21/c 中结晶,晶胞参数 a = 10.6345(3) A, b = 14.7074(4) A, c = 10.6677(4) A, β = 101.453(2)635°和 V27 (9) A3。在该化合物中,六氢嘧啶环(C7/C8/C12/N1/C14/N2)采用沙发构象。起皱参数为 Q = 0.5117A、Ɵ = 129.59° 和 φ =304.0802°。Hirshfeld 表面的分析表明存在氢键 C-H•••F、C-H•••O、O-H•••O、N-H•••O 和 π•••π 堆积稳定系统。能量优化结构是使用密度泛函理论 (DFT) 计算的,并由混合泛函理论 (B3LYP) 在 Gaussian09_revision C0 中的 6-311++G(d,p) 基组中采用。1个程序包。自然键轨道分析结果表明,最高能量转移发生在供体 σ(O4-H43) 和受体 σ*(C34-H36) 之间,通过羧酸盐链达到 604.21 kJ/mol。此外,振动分配、全局反应性描述符、能隙和分子静电势 (MEP) 也清楚地了解分子的化学和生物活性。合成化合物的第一个超极化值 7.2987 × 10-30 esu 是尿素(0.3728 × 10-30 esu)的 20 倍。通过时间相关密度泛函理论(TD-DFT)计算计算合成晶体的单电子激发,并使用记录的紫外光谱进行比较研究。配备 532 nm 连续 Nd 的单光束 Z 扫描单元:用5ns脉冲宽度的YAG波激光测量三阶非线性光学性质表明所研究的分子具有有效的二光子吸收,有效吸收系数更高。三阶磁化率值的虚部和实部分别确定为 Im χ3 = 2.31 × 10−6 cm W−1 和 Re χ3 = 8.74 × 10−8 cm2 W−1。
更新日期:2021-03-01
中文翻译:
新型氟化六氢嘧啶的合成、晶体结构、Hirshfeld 表面、DFT 计算、Z 扫描和非线性光学研究
摘要 合成了一种新的氟化六氢嘧啶,并利用单晶X射线衍射技术阐明了其晶体结构。该化合物 C15H17F3N2O6 在单斜空间群 P21/c 中结晶,晶胞参数 a = 10.6345(3) A, b = 14.7074(4) A, c = 10.6677(4) A, β = 101.453(2)635°和 V27 (9) A3。在该化合物中,六氢嘧啶环(C7/C8/C12/N1/C14/N2)采用沙发构象。起皱参数为 Q = 0.5117A、Ɵ = 129.59° 和 φ =304.0802°。Hirshfeld 表面的分析表明存在氢键 C-H•••F、C-H•••O、O-H•••O、N-H•••O 和 π•••π 堆积稳定系统。能量优化结构是使用密度泛函理论 (DFT) 计算的,并由混合泛函理论 (B3LYP) 在 Gaussian09_revision C0 中的 6-311++G(d,p) 基组中采用。1个程序包。自然键轨道分析结果表明,最高能量转移发生在供体 σ(O4-H43) 和受体 σ*(C34-H36) 之间,通过羧酸盐链达到 604.21 kJ/mol。此外,振动分配、全局反应性描述符、能隙和分子静电势 (MEP) 也清楚地了解分子的化学和生物活性。合成化合物的第一个超极化值 7.2987 × 10-30 esu 是尿素(0.3728 × 10-30 esu)的 20 倍。通过时间相关密度泛函理论(TD-DFT)计算计算合成晶体的单电子激发,并使用记录的紫外光谱进行比较研究。配备 532 nm 连续 Nd 的单光束 Z 扫描单元:用5ns脉冲宽度的YAG波激光测量三阶非线性光学性质表明所研究的分子具有有效的二光子吸收,有效吸收系数更高。三阶磁化率值的虚部和实部分别确定为 Im χ3 = 2.31 × 10−6 cm W−1 和 Re χ3 = 8.74 × 10−8 cm2 W−1。






























 京公网安备 11010802027423号
京公网安备 11010802027423号