当前位置:
X-MOL 学术
›
J. Mol. Struct.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
An analysis of structural, spectroscopic, quantum chemical and in silico studies of ethyl 3-[(pyridin-2-yl)amino]propanoate: A potential thrombin inhibitor
Journal of Molecular Structure ( IF 4.0 ) Pub Date : 2021-02-01 , DOI: 10.1016/j.molstruc.2020.129378 Nuthalapati Poojith , Nannapaneni Usha Rani , Krishna Murthy Potla , J. John Rose , P.A. Suchetan , Renjith Raveendran Pillai , Suneetha Vankayalapati
Journal of Molecular Structure ( IF 4.0 ) Pub Date : 2021-02-01 , DOI: 10.1016/j.molstruc.2020.129378 Nuthalapati Poojith , Nannapaneni Usha Rani , Krishna Murthy Potla , J. John Rose , P.A. Suchetan , Renjith Raveendran Pillai , Suneetha Vankayalapati
|
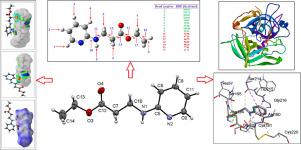
Abstract In the present investigation, we report an analysis of the structural, spectroscopic characterization, reactivity parameters, topological studies, and molecular docking studies of the synthesized ethyl 3-[(pyridin-2-yl)amino]propanoate (abbreviated to EPYAPP), C10H14N2O2. The molecular and crystal structure was determined by using the single-crystal X-ray diffraction technique. The whole molecule is planar with r.m.s.d. of all the non-hydrogen atoms being 0.041(2) A. Further, the dihedral angle between the planes of the aromatic ring and the side chain is 1.6(1)o. The crystal structure features a pair of N-H…N hydrogen-bonded dimers connected via two C-H...π (π electrons of the aromatic ring) interactions to form a two-dimensional zig-zag sheet propagating parallel to the bc plane. The qualitative and quantitative estimation of close contacts in the solid phase of the title compound was performed through the 3D-Hirshfeld surface analysis and 2D-finger print plots. The quantum chemical calculations of the EPYAPP compound were performed at DFT/B3LYP/6-311++G(d,p) method at the ground state in the gas phase. The detailed investigation of each vibrational wave number was carried out by using the VEDA4 package, and theoretical results showed an excellent mutual agreement with the experimental spectral data. The HOMO-LUMO orbital energy calculations, chemical reactivity descriptors, and natural bond orbital analysis were also performed. Prone reactive sites of the title compound have been identified by the molecular electrostatic surface potential and Fukui functions, which are mapped to the electron density surfaces. Hydrogen bond dissociation energies and bond dissociation energies for all other single bonds were further calculated for the EPYAPP molecule in order to examine the autoxidation mechanism and degradation properties. The title compound forms a stable complex with human alpha thrombin (PDB code: 1PPB) (binding energy -7.03 kcal/mol)) antagonist and could be a lead compound for developing new thrombin inhibitor or anti-thrombotic drugs.
中文翻译:

3-[(吡啶-2-基)氨基]丙酸乙酯的结构、光谱、量子化学和计算机研究分析:一种潜在的凝血酶抑制剂
摘要 在本研究中,我们报告了对合成的 3-[(pyridin-2-yl)amino] 丙酸乙酯(缩写为 EPYAPP)的结构、光谱表征、反应参数、拓扑研究和分子对接研究的分析, C10H14N2O2。分子和晶体结构是通过使用单晶 X 射线衍射技术确定的。整个分子是平面的,所有非氢原子的rmsd为0.041(2)A。此外,芳环平面与侧链平面之间的二面角为1.6(1)o。晶体结构的特点是一对 NH…N 氢键二聚体通过两个 CH…π(芳环的 π 电子)相互作用连接,形成平行于 bc 平面传播的二维锯齿形片。通过 3D-Hirshfeld 表面分析和 2D 指纹图对标题化合物固相中的密切接触进行定性和定量估计。EPYAPP 化合物的量子化学计算是在气相基态下以 DFT/B3LYP/6-311++G(d,p) 方法进行的。使用VEDA4软件包对每个振动波数进行了详细研究,理论结果与实验光谱数据相互吻合。还进行了 HOMO-LUMO 轨道能量计算、化学反应性描述符和自然键轨道分析。标题化合物的易反应位点已通过分子静电表面电位和 Fukui 函数确定,它们映射到电子密度表面。进一步计算了 EPYAPP 分子的氢键解离能和所有其他单键的键解离能,以检查自氧化机制和降解特性。标题化合物与人 α 凝血酶(PDB 代码:1PPB)(结合能 -7.03 kcal/mol))拮抗剂形成稳定的复合物,可作为开发新型凝血酶抑制剂或抗血栓药物的先导化合物。
更新日期:2021-02-01
中文翻译:
3-[(吡啶-2-基)氨基]丙酸乙酯的结构、光谱、量子化学和计算机研究分析:一种潜在的凝血酶抑制剂
摘要 在本研究中,我们报告了对合成的 3-[(pyridin-2-yl)amino] 丙酸乙酯(缩写为 EPYAPP)的结构、光谱表征、反应参数、拓扑研究和分子对接研究的分析, C10H14N2O2。分子和晶体结构是通过使用单晶 X 射线衍射技术确定的。整个分子是平面的,所有非氢原子的rmsd为0.041(2)A。此外,芳环平面与侧链平面之间的二面角为1.6(1)o。晶体结构的特点是一对 NH…N 氢键二聚体通过两个 CH…π(芳环的 π 电子)相互作用连接,形成平行于 bc 平面传播的二维锯齿形片。通过 3D-Hirshfeld 表面分析和 2D 指纹图对标题化合物固相中的密切接触进行定性和定量估计。EPYAPP 化合物的量子化学计算是在气相基态下以 DFT/B3LYP/6-311++G(d,p) 方法进行的。使用VEDA4软件包对每个振动波数进行了详细研究,理论结果与实验光谱数据相互吻合。还进行了 HOMO-LUMO 轨道能量计算、化学反应性描述符和自然键轨道分析。标题化合物的易反应位点已通过分子静电表面电位和 Fukui 函数确定,它们映射到电子密度表面。进一步计算了 EPYAPP 分子的氢键解离能和所有其他单键的键解离能,以检查自氧化机制和降解特性。标题化合物与人 α 凝血酶(PDB 代码:1PPB)(结合能 -7.03 kcal/mol))拮抗剂形成稳定的复合物,可作为开发新型凝血酶抑制剂或抗血栓药物的先导化合物。





























 京公网安备 11010802027423号
京公网安备 11010802027423号