当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
MOF-808中Pt团簇稳定性的密度泛函理论研究
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2020-09-28 , DOI: 10.1039/d0cp04444j Xiaohui Song 1, 2, 3, 4 , Donghai Mei 1, 2, 3, 4
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2020-09-28 , DOI: 10.1039/d0cp04444j Xiaohui Song 1, 2, 3, 4 , Donghai Mei 1, 2, 3, 4
Affiliation
|
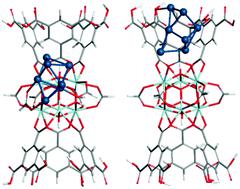
由于金属团簇的几何和电子性质,金属有机骨架(MOF)封装的金属团簇已显示出优异的催化活性,这在很大程度上取决于吸附位点以及封装的金属团簇的大小和形态。在目前的工作中,使用密度泛函理论计算和从头算分子动力学模拟研究了MOF-808框架结构上Pt n(n = 1-23)簇的锚定位,稳定性和团聚概率。已经发现,Pt n(n = 1–7)团簇在Zr 6处的结合更牢固金属节点站点比在接口站点和链接器站点要多。吸附后,大量电子(+0.92至+1.96 | e |)从Pt n团簇转移至MOF骨架。在Zr 6金属节点上单个Pt 1原子的团聚不可能形成Pt n簇,而在界面或连接体上的团聚在能量上是可行的。与单个Zr 6节点相比,Pt n簇与两个Zr 6金属节点的键合更弱,电子更少(+0.12至+0.89 | e|)转移。最后,我们的计算表明,在单个Pt原子上的CO吸附在界面处稳定,从而防止了它与两个Zr 6金属节点之间的Pt n团簇进一步聚集。

"点击查看英文标题和摘要"
更新日期:2020-10-15
"点击查看英文标题和摘要"





























 京公网安备 11010802027423号
京公网安备 11010802027423号