当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Enantiomeric Transitions in the Chiral Cluster Au15 Studied by a Reaction Coordinate Deduced from Molecular Dynamics Simulations
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2020-09-28 , DOI: 10.1021/acs.jpca.0c05099 Chong Chiat Lim, San Kiong Lai
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2020-09-28 , DOI: 10.1021/acs.jpca.0c05099 Chong Chiat Lim, San Kiong Lai
|
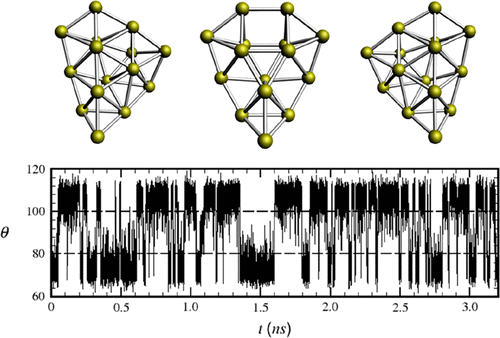
A recently developed modified basin hopping (MBH) optimization algorithm, combined with an energy function calculated by the semiempirical density functional tight-binding (DFTB) theory, was applied to determine the lowest-energy structures of Aun clusters with size n = 3–20. It was predicted from the DFTB/MBH optimization algorithm calculations that clusters Au10, Au15, and Au18 exhibit chiral properties; i.e., each of these three clusters possesses the same energy value and associated with it are two nonsuperposable mirror-image clusters. In the potential energy landscape, there thus exist multidimensional barriers separating the two enantiomers, and this lowest-energy double-well morphology is surrounded by potential-energy minima of higher energies. In this paper, we have chosen to study the chiral cluster Au15 by employing an isothermal Brownian-type molecular dynamics simulation to discern in greater detail its conformational transition from one enantiomer, say left, to its right counterpart. To facilitate our analysis of the simulation data, we transpose the multidimensional configurational space description to a lower dimensional collective variable (CV) space spanned by two geometry-relevant CVs. The thermally driven progression and mechanism of enantiomeric transitions between the left and right enantiomers will be our main focus, and the strategy is to dissect the time development of the CVs collected from different sets of independent simulation runs. From simulation data, we found that an understanding of the dynamics of enantiomeric transitions needs first to seek out the left and right enantiomers through a molecular modeling and visualizing program, then to ferret out and identify between the left and right enantiomers a symmetrical structure, and finally to define from the latter a reaction coordinate. We showed in this work that this single reaction coordinate is predictive in unraveling the left ⇌ right enantiomeric transition events, providing a specific inkling of the transition time span and its associated distribution which can be checked further for its reasonableness by the autocorrelation function and a vibrational analysis, all of which shed light on the mechanisms of transition.
中文翻译:

通过分子动力学模拟推导的反应坐标研究手性簇Au 15中的对映体跃迁
最近开发的一种改进的盆地跳跃(MBH)优化算法,结合通过半经验密度泛函紧束缚(DFTB)理论计算的能量函数,确定了n = 3–的Au n团簇的最低能量结构。20 根据DFTB / MBH优化算法计算预测,该算法将Au 10,Au 15和Au 18聚类表现出手性 即,这三个簇中的每一个都具有相同的能量值,并且与之相关联的是两个不可叠加的镜像簇。因此,在势能图中,存在将两个对映异构体分开的多维壁垒,这种最低能量的双阱形态被较高能量的势能最小值所包围。在本文中,我们选择研究手性团簇Au 15通过使用等温布朗型分子动力学模拟,更详细地识别其从一个对映异构体(如左图)向右对映异构体的构象转变。为了方便我们对仿真数据的分析,我们将多维配置空间描述转换为由两个与几何相关的CV跨越的低维集体变量(CV)空间。左右对映异构体之间的对映体转变的热驱动过程和机理将是我们的主要研究重点,其策略是剖析从不同组独立模拟运行中收集的CV的时间发展。根据仿真数据,我们发现,了解对映体转变的动力学首先需要通过分子建模和可视化程序找出左右对映体,然后找出并确定左右对映体之间的对称结构,最后从后者是反应坐标。我们在这项工作中表明,该单一反应坐标可预测解开左⇌右对映体转变事件,提供了转变时间跨度及其相关分布的特定信息,可以通过自相关函数和振动进一步检查其合理性。分析,所有这些都阐明了转变的机制。最后从后者定义一个反应坐标。我们在这项工作中表明,该单一反应坐标可预测解开左⇌右对映体转变事件,提供了转变时间跨度及其相关分布的特定信息,可以通过自相关函数和振动进一步检查其合理性。分析,所有这些都阐明了转变的机制。最后从后者定义一个反应坐标。我们在这项工作中表明,该单一反应坐标可预测解开左⇌右对映体转变事件,提供了转变时间跨度及其相关分布的特定信息,可以通过自相关函数和振动进一步检查其合理性。分析,所有这些都阐明了转变的机制。
更新日期:2020-10-22
中文翻译:
通过分子动力学模拟推导的反应坐标研究手性簇Au 15中的对映体跃迁
最近开发的一种改进的盆地跳跃(MBH)优化算法,结合通过半经验密度泛函紧束缚(DFTB)理论计算的能量函数,确定了n = 3–的Au n团簇的最低能量结构。20 根据DFTB / MBH优化算法计算预测,该算法将Au 10,Au 15和Au 18聚类表现出手性 即,这三个簇中的每一个都具有相同的能量值,并且与之相关联的是两个不可叠加的镜像簇。因此,在势能图中,存在将两个对映异构体分开的多维壁垒,这种最低能量的双阱形态被较高能量的势能最小值所包围。在本文中,我们选择研究手性团簇Au 15通过使用等温布朗型分子动力学模拟,更详细地识别其从一个对映异构体(如左图)向右对映异构体的构象转变。为了方便我们对仿真数据的分析,我们将多维配置空间描述转换为由两个与几何相关的CV跨越的低维集体变量(CV)空间。左右对映异构体之间的对映体转变的热驱动过程和机理将是我们的主要研究重点,其策略是剖析从不同组独立模拟运行中收集的CV的时间发展。根据仿真数据,我们发现,了解对映体转变的动力学首先需要通过分子建模和可视化程序找出左右对映体,然后找出并确定左右对映体之间的对称结构,最后从后者是反应坐标。我们在这项工作中表明,该单一反应坐标可预测解开左⇌右对映体转变事件,提供了转变时间跨度及其相关分布的特定信息,可以通过自相关函数和振动进一步检查其合理性。分析,所有这些都阐明了转变的机制。最后从后者定义一个反应坐标。我们在这项工作中表明,该单一反应坐标可预测解开左⇌右对映体转变事件,提供了转变时间跨度及其相关分布的特定信息,可以通过自相关函数和振动进一步检查其合理性。分析,所有这些都阐明了转变的机制。最后从后者定义一个反应坐标。我们在这项工作中表明,该单一反应坐标可预测解开左⇌右对映体转变事件,提供了转变时间跨度及其相关分布的特定信息,可以通过自相关函数和振动进一步检查其合理性。分析,所有这些都阐明了转变的机制。












































 京公网安备 11010802027423号
京公网安备 11010802027423号