Journal of Molecular Modeling ( IF 2.1 ) Pub Date : 2020-09-26 , DOI: 10.1007/s00894-020-04556-5 Mohsen Doust Mohammadi 1 , Hewa Y Abdullah 2
|
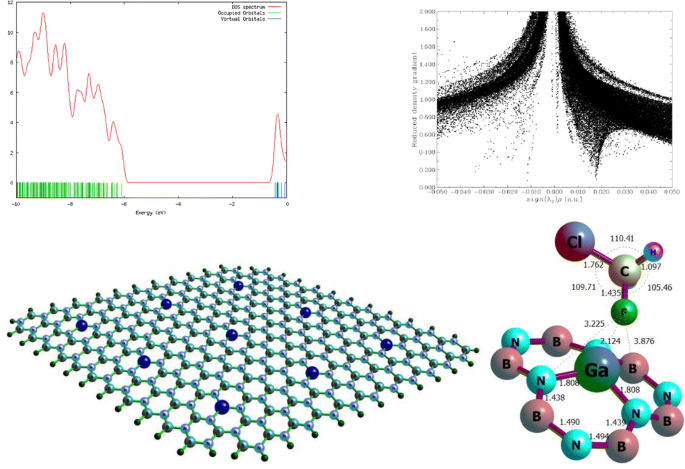
In the present investigation, the feasibility of detecting the chlorofluoromethane (CFM) gas molecule onto the outer surface of pristine single layer boron nitride nanosheet (BNNS), as well as its aluminum (Al)– and gallium (Ga)–doped structures, was carefully evaluated. For achieving this goal, a density functional theory level of study using the Perdew, Burke, and Ernzerhof exchange–correlation (PBEPBE) functional together with a 6-311G(d) basis set has been used. Subsequently, the B3LYP, CAM-B3LYP, wB97XD, and M062X functionals with a 6-311G(d) basis set were also employed to consider the single-point energies. Natural bond orbital (NBO) and quantum theory of atoms in molecules (QTAIM) were implemented by using the B3LYP-D3/6-311G(d) method, and the results were compatible with the electronic properties. In this regard, the total density of states (TDOSs), the Wiberg bond index (WBI), natural charge, natural electron configuration, donor–acceptor natural bond orbital interactions, and the second-order perturbation energies are performed to explore the nature of the intermolecular interactions. All of the energy calculations and population analyses denote that by adsorbing of the gas molecule onto the surface of the considered nanostructures, the intermolecular interactions are of the type of strong chemical adsorption. Among the doped nanosheets, Ga-doped nanosheet has very high adsorption energy compared with other elements (i.e., Ga-doped > Al-doped > pristine). Generally, it was revealed that the sensitivity of the adsorption will be increased when the gas molecule interacts with decorated nanosheets and decrease the HOMO-LUMO band gap; therefore, the change of electronic properties can be used to design suitable nanosensors to detect CFM gas.
中文翻译:
DFT,NBO和QTAIM研究在原始,以及Al和Ga掺杂的氮化硼纳米片上的吸附氯氟甲烷。
在本研究中,检测原始的单层氮化硼纳米片(BNNS)以及铝(Al)和镓(Ga)掺杂结构外表面上的氯氟甲烷(CFM)气体分子的可行性是仔细评估。为了实现这一目标,使用了Perdew,Burke和Ernzerhof交换相关(PBEPBE)泛函以及6-311G(d)基础集的密度泛函理论研究水平。随后,还使用具有6-311G(d)基集的B3LYP,CAM-B3LYP,wB97XD和M062X功能来考虑单点能量。利用B3LYP-D3 / 6-311G(d)方法实现了自然键轨道(NBO)和分子原子量子理论(QTAIM),其结果与电子性质兼容。在这方面,进行了状态总密度(TDOS),Wiberg键指数(WBI),自然电荷,自然电子构型,供体-受体自然键轨道相互作用以及二阶微扰能的研究,以探索分子间相互作用的性质。所有的能量计算和总体分析表明,通过将气体分子吸附到所考虑的纳米结构的表面上,分子间的相互作用属于强化学吸附类型。在掺杂的纳米片中,与其他元素(即,Ga掺杂> Al掺杂>原始)相比,Ga掺杂的纳米片具有非常高的吸附能。一般地,揭示了当气体分子与装饰的纳米片相互作用时吸附的敏感性将增加并且减小HOMO-LUMO带隙。因此,




















































 京公网安备 11010802027423号
京公网安备 11010802027423号