当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Role of Reduced CeO2(110) Surface for CO2 Reduction to CO and Methanol
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2016-07-18 00:00:00 , DOI: 10.1021/acs.jpcc.6b02860 Neetu Kumari 1 , M. Ali Haider 1 , Manish Agarwal 2 , Nishant Sinha 3 , Suddhasatwa Basu 1
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2016-07-18 00:00:00 , DOI: 10.1021/acs.jpcc.6b02860 Neetu Kumari 1 , M. Ali Haider 1 , Manish Agarwal 2 , Nishant Sinha 3 , Suddhasatwa Basu 1
Affiliation
|
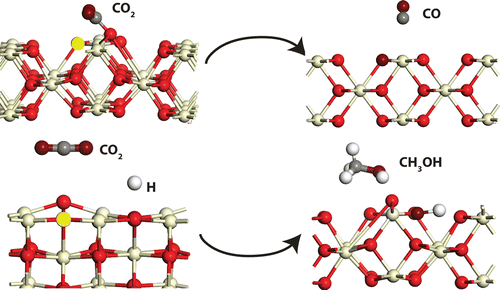
Density functional theory (DFT) calculations were performed to study the mechanism of carbon dioxide (CO2) reduction to carbon monoxide (CO) and methanol (CH3OH) on CeO2(110) surface. CO2 dissociates to CO on interacting with the oxygen vacancy on reduced ceria surface. The oxygen atom heals the vacancy site and regenerates the stoichiometric surface via a redox mechanism with intrinsic activation and reaction energies of 259.2 and 238.6 kJ/mol, respectively. Lateral interaction of oxygen vacancies were studied by the generation of two oxygen vacancies per unit of CeO2 surface. Compared to a single isolated vacancy, the activation and reaction energies of CO2 dissociation on a divacancy were approximately reduced to half of its value. Hydrogen atom coadsorbed on the surface was observed to assist CO2 dissociation by forming a carboxyl intermediate, CO2+H → COOH (ΔEact = 39.0 kJ/mol, ΔH = −69.2 kJ/mol) which on subsequent dissociation produces CO via the redox mechanism. On hydrogenation, CO is likely to produce methanol. The energetics of CO hydrogenation to produce methanol showed exothermic steps with activation barriers comparable to the DFT calculations reported for Cu (111) and CeO2–x/Cu(111) interface. While on the stoichiometric surface, COOH dissociation COOH → CO+OH (ΔEact = 55.6 kJ/mol, ΔH = 5.7 kJ/mol) is likely to be difficult as compared to rest of the elementary steps, whereas on the reduced surface the energetics of the same step were significantly lowered (ΔEact = 47.4 kJ/mol, ΔH = −80.4 kJ/mol). In comparison, hydrogenation of methoxy, H3CO+H → H3COH, appears to be relatively difficult (ΔEact = 58.7 kJ/mol) on the reduced surface.
中文翻译:

还原的CeO 2(110)表面在将CO 2还原为CO和甲醇中的作用
进行了密度泛函理论(DFT)计算,以研究二氧化碳(CO 2)在CeO 2(110)表面还原为一氧化碳(CO)和甲醇(CH 3 OH )的机理。CO 2与二氧化铈表面还原后的氧空位相互作用时解离为CO。氧原子可以通过固有的活化能和反应能分别为259.2和238.6 kJ / mol的氧化还原机理修复空位并再生化学计量表面。通过每单位CeO 2表面产生两个氧空位来研究氧空位的横向相互作用。与单个空位相比,CO 2的活化能和反应能空缺的解离大约减少到其价值的一半。观察到共吸附在表面上的氢原子通过形成羧基中间体CO 2 + H→COOH(ΔE act = 39.0 kJ / mol,ΔH = -69.2 kJ / mol)协助CO 2离解,该羧基中间体在随后的离解中会生成CO通过氧化还原机制。氢化时,一氧化碳很可能产生甲醇。CO加氢生成甲醇的能量学显示出放热步骤,其活化势垒可与针对Cu(111)和CeO 2– x / Cu(111)界面报道的DFT计算相媲美。在化学计量表面上,COOH分解为COOH→CO + OH(ΔE act = 55.6 kJ / mol,Δ与其余基本步骤相比,H = 5.7 kJ / mol)可能很困难,而在减小的表面上,同一步骤的能量显着降低(ΔE act = 47.4 kJ / mol,ΔH = -80.4) kJ / mol)。相比之下,在还原的表面上,甲氧基H 3 CO + H→H 3 COH的氢化似乎相对困难(ΔE act = 58.7 kJ / mol)。
更新日期:2016-07-18
中文翻译:
还原的CeO 2(110)表面在将CO 2还原为CO和甲醇中的作用
进行了密度泛函理论(DFT)计算,以研究二氧化碳(CO 2)在CeO 2(110)表面还原为一氧化碳(CO)和甲醇(CH 3 OH )的机理。CO 2与二氧化铈表面还原后的氧空位相互作用时解离为CO。氧原子可以通过固有的活化能和反应能分别为259.2和238.6 kJ / mol的氧化还原机理修复空位并再生化学计量表面。通过每单位CeO 2表面产生两个氧空位来研究氧空位的横向相互作用。与单个空位相比,CO 2的活化能和反应能空缺的解离大约减少到其价值的一半。观察到共吸附在表面上的氢原子通过形成羧基中间体CO 2 + H→COOH(ΔE act = 39.0 kJ / mol,ΔH = -69.2 kJ / mol)协助CO 2离解,该羧基中间体在随后的离解中会生成CO通过氧化还原机制。氢化时,一氧化碳很可能产生甲醇。CO加氢生成甲醇的能量学显示出放热步骤,其活化势垒可与针对Cu(111)和CeO 2– x / Cu(111)界面报道的DFT计算相媲美。在化学计量表面上,COOH分解为COOH→CO + OH(ΔE act = 55.6 kJ / mol,Δ与其余基本步骤相比,H = 5.7 kJ / mol)可能很困难,而在减小的表面上,同一步骤的能量显着降低(ΔE act = 47.4 kJ / mol,ΔH = -80.4) kJ / mol)。相比之下,在还原的表面上,甲氧基H 3 CO + H→H 3 COH的氢化似乎相对困难(ΔE act = 58.7 kJ / mol)。






























 京公网安备 11010802027423号
京公网安备 11010802027423号