Medicinal Chemistry ( IF 1.9 ) Pub Date : 2021-07-31 , DOI: 10.2174/1573406416666200420081540 Tran Khac Vu 1 , Nguyen Thi Thanh 1 , Nguyen Van Minh 1 , Nguyen Huong Linh 1 , Nguyen Thi Phương Thao 1 , Trương Thuc Bao Nguyen 1 , Doan Thi Hien 1 , Luu Van Chinh 2 , Ta Hong Duc 1 , Lai Duc Anh 3 , Pham-The Hai 3
|
Background: The target-based approach to drug discovery currently attracts a great deal of interest from medicinal chemists in anticancer drug discovery and development. Histone deacetylase (HDAC) inhibitors represent an extensive class of targeted anti-cancer agents. Among the most explored structure moieties, hydroxybenzamides and hydroxypropenamides have been demonstrated to have potential HDAC inhibitory effects. Several compounds of these structural classes have been approved for clinical uses to treat different types of cancer, such as vorinostat and belinostat.
Aims: This study aims at developing novel HDAC inhibitors bearing conjugated quinazolinone scaffolds with potential cytotoxicity against different cancer cell lines.
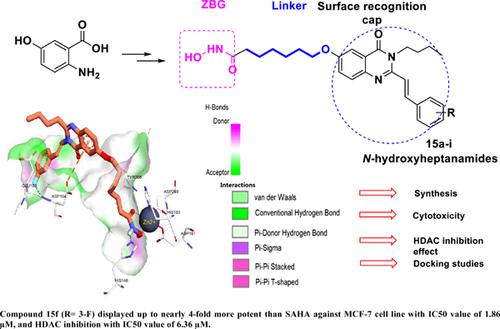
Methods: A series of novel N-hydroxyheptanamides incorporating conjugated 6-hydroxy-2 methylquinazolin- 4(3H)-ones (15a-l) was designed, synthesized and evaluated for HDAC inhibitory potency as well as cytotoxicity against three human cancer cell lines, including HepG-2, MCF-7 and SKLu-1. Molecular simulations were finally performed to gain more insight into the structureactivity relationships.
Results: It was found that among novel conjugated quinazolinone-based hydroxamic acids synthesized, compounds 15a, 15c and 15f were the most potent, both in terms of HDAC inhibition and cytotoxicity. Especially, compound 15f displayed up to nearly 4-fold more potent than SAHA (vorinostat) in terms of cytotoxicity against MCF-7 cell line with IC50 value of 1.86 μM, and HDAC inhibition with IC50 value of 6.36 μM. Docking experiments on HDAC2 isozyme showed that these compounds bound to HDAC2 with binding affinities ranging from -10.08 to -14.93 kcal/mol compared to SAHA (-15.84 kcal/mol). It was also found in this research that most of the target compounds seemed to be more cytotoxic toward SKLu-1than MCF-7 and HepG-2.
Conclusion: The resesrch results suggest that some hydroxamic acids could emerge for further evaluation and the results are well served as basics for further design of more potent HDAC inhibitors and antitumor agents.
中文翻译:
新型共轭喹唑啉酮基异羟肟酸:设计、合成和生物学评价
背景:基于靶标的药物发现方法目前在抗癌药物的发现和开发中引起了药物化学家的极大兴趣。组蛋白去乙酰化酶 (HDAC) 抑制剂代表了一类广泛的靶向抗癌药物。在研究最多的结构部分中,羟基苯甲酰胺和羟基丙烯酰胺已被证明具有潜在的 HDAC 抑制作用。这些结构类别的几种化合物已被批准用于临床治疗不同类型的癌症,例如伏立诺他和贝立诺他。
目的:本研究旨在开发新型 HDAC 抑制剂,该抑制剂带有对不同癌细胞系具有潜在细胞毒性的共轭喹唑啉酮支架。
方法:设计、合成了一系列结合了共轭 6-羟基-2 甲基喹唑啉-4(3H)-酮 (15a-l) 的新型 N-羟基庚酰胺,并评估了 HDAC 抑制效力以及对三种人类癌细胞系的细胞毒性,包括 HepG-2、MCF-7 和 SKLu-1。最后进行分子模拟以更深入地了解构效关系。
结果:发现在合成的新型共轭喹唑啉酮异羟肟酸中,化合物 15a、15c 和 15f 在 HDAC 抑制和细胞毒性方面最有效。特别是,化合物 15f 在对 MCF-7 细胞系的细胞毒性方面表现出比 SAHA(伏立诺他)高近 4 倍,IC 50值为 1.86 μM,HDAC 抑制作用的 IC 50值为 6.36 μM。HDAC2 同工酶的对接实验表明,与 SAHA (-15.84 kcal/mol) 相比,这些化合物以 -10.08 至 -14.93 kcal/mol 的结合亲和力与 HDAC2 结合。本研究还发现,大多数目标化合物对 SKLu-1 的细胞毒性似乎比 MCF-7 和 HepG-2 更强。
结论:研究结果表明,一些异羟肟酸可能出现用于进一步评估,结果很好地为进一步设计更有效的 HDAC 抑制剂和抗肿瘤药物奠定了基础。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号