当前位置:
X-MOL 学术
›
Comp. Mater. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Physically inspired atom-centered symmetry functions for the construction of high dimensional neural network potential energy surfaces
Computational Materials Science ( IF 3.1 ) Pub Date : 2021-01-01 , DOI: 10.1016/j.commatsci.2020.110071 Kangyu Zhang , Lichang Yin , Gang Liu
Computational Materials Science ( IF 3.1 ) Pub Date : 2021-01-01 , DOI: 10.1016/j.commatsci.2020.110071 Kangyu Zhang , Lichang Yin , Gang Liu
|
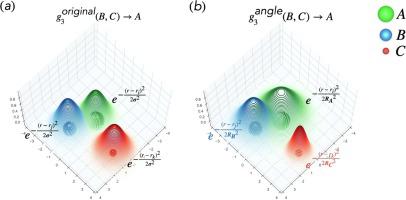
Abstract Among different atomistic neural network (AtNN) potential energy surfaces (PESs), the Behler-Parrinello neural network (BPNN) based on atom-centered symmetry functions (ACSFs) has been proved to be capable of constructing accurate PESs for various crystals. A judicious setting of the parameters of the ACSFs largely determines the accuracy of a BPNN PES. However, this is typically an ad hoc and tedious task requiring highly acute chemical intuition. To address this issue, we derived a set of physically inspired ACSFs from the effective densities of atoms, in which the radii of atoms are naturally incorporated. Therefore, the parameters of the physically inspired ACSFs can be directly chosen based on the types of chemical bonds within a target system. Compared with the original ones, the physically inspired ACSFs are more suitable for complex systems based on its better performance on predicting the formation enthalpies of molecules in QM9 database. Moreover, the physically inspired ACSFs can also effectively accelerate the convergence of the atomic forces during the training of an AtNN PES. With the physically inspired ACSFs, we constructed a highly accurate AtNN PES for a solid electrolyte Li10GeP2S12. Based on the AtNN PES, we studied the bulk Li ion diffusion within Li10GeP2S12 by molecular dynamics (MD) simulations. The MD results well reproduced the experimental results, indicating the high accuracy of the AtNN PES constructed with the physically inspired ACSFs.
中文翻译:

用于构建高维神经网络势能面的物理启发的以原子为中心的对称函数
摘要 在不同的原子神经网络 (AtNN) 势能面 (PES) 中,基于原子中心对称函数 (ACSF) 的 Behler-Parrinello 神经网络 (BPNN) 已被证明能够为各种晶体构建准确的 PES。ACSF 参数的明智设置在很大程度上决定了 BPNN PES 的准确性。然而,这通常是一项需要高度敏锐的化学直觉的临时且乏味的任务。为了解决这个问题,我们从原子的有效密度中衍生出一组受物理启发的 ACSF,其中原子的半径是自然结合的。因此,可以根据目标系统内的化学键类型直接选择物理启发的 ACSF 的参数。与原版相比,基于物理启发的 ACSF 在预测 QM9 数据库中分子的形成焓方面具有更好的性能,因此更适合复杂系统。此外,在 AtNN PES 的训练过程中,受物理启发的 ACSF 还可以有效地加速原子力的收敛。借助物理启发的 ACSF,我们为固体电解质 Li10GeP2S12 构建了高度准确的 AtNN PES。基于 AtNN PES,我们通过分子动力学 (MD) 模拟研究了 Li10GeP2S12 内的体锂离子扩散。MD 结果很好地再现了实验结果,表明使用受物理启发的 ACSF 构建的 AtNN PES 具有很高的准确性。在 AtNN PES 的训练过程中,受物理启发的 ACSF 还可以有效地加速原子力的收敛。借助物理启发的 ACSF,我们为固体电解质 Li10GeP2S12 构建了高度准确的 AtNN PES。基于 AtNN PES,我们通过分子动力学 (MD) 模拟研究了 Li10GeP2S12 内的体锂离子扩散。MD 结果很好地再现了实验结果,表明使用受物理启发的 ACSF 构建的 AtNN PES 具有很高的准确性。在 AtNN PES 的训练过程中,受物理启发的 ACSF 还可以有效地加速原子力的收敛。借助物理启发的 ACSF,我们为固体电解质 Li10GeP2S12 构建了高度准确的 AtNN PES。基于 AtNN PES,我们通过分子动力学 (MD) 模拟研究了 Li10GeP2S12 内的体锂离子扩散。MD 结果很好地再现了实验结果,表明使用受物理启发的 ACSF 构建的 AtNN PES 具有很高的准确性。
更新日期:2021-01-01
中文翻译:
用于构建高维神经网络势能面的物理启发的以原子为中心的对称函数
摘要 在不同的原子神经网络 (AtNN) 势能面 (PES) 中,基于原子中心对称函数 (ACSF) 的 Behler-Parrinello 神经网络 (BPNN) 已被证明能够为各种晶体构建准确的 PES。ACSF 参数的明智设置在很大程度上决定了 BPNN PES 的准确性。然而,这通常是一项需要高度敏锐的化学直觉的临时且乏味的任务。为了解决这个问题,我们从原子的有效密度中衍生出一组受物理启发的 ACSF,其中原子的半径是自然结合的。因此,可以根据目标系统内的化学键类型直接选择物理启发的 ACSF 的参数。与原版相比,基于物理启发的 ACSF 在预测 QM9 数据库中分子的形成焓方面具有更好的性能,因此更适合复杂系统。此外,在 AtNN PES 的训练过程中,受物理启发的 ACSF 还可以有效地加速原子力的收敛。借助物理启发的 ACSF,我们为固体电解质 Li10GeP2S12 构建了高度准确的 AtNN PES。基于 AtNN PES,我们通过分子动力学 (MD) 模拟研究了 Li10GeP2S12 内的体锂离子扩散。MD 结果很好地再现了实验结果,表明使用受物理启发的 ACSF 构建的 AtNN PES 具有很高的准确性。在 AtNN PES 的训练过程中,受物理启发的 ACSF 还可以有效地加速原子力的收敛。借助物理启发的 ACSF,我们为固体电解质 Li10GeP2S12 构建了高度准确的 AtNN PES。基于 AtNN PES,我们通过分子动力学 (MD) 模拟研究了 Li10GeP2S12 内的体锂离子扩散。MD 结果很好地再现了实验结果,表明使用受物理启发的 ACSF 构建的 AtNN PES 具有很高的准确性。在 AtNN PES 的训练过程中,受物理启发的 ACSF 还可以有效地加速原子力的收敛。借助物理启发的 ACSF,我们为固体电解质 Li10GeP2S12 构建了高度准确的 AtNN PES。基于 AtNN PES,我们通过分子动力学 (MD) 模拟研究了 Li10GeP2S12 内的体锂离子扩散。MD 结果很好地再现了实验结果,表明使用受物理启发的 ACSF 构建的 AtNN PES 具有很高的准确性。






























 京公网安备 11010802027423号
京公网安备 11010802027423号