当前位置:
X-MOL 学术
›
Appl. Catal. A Gen.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
从密度泛函理论计算中H2,O2,H2O,CO2,CO,CH4和C2Hx在金属Mo2C(001)和Mo / C终止的Mo2C(101)上的活化机理
Applied Catalysis A: General ( IF 4.7 ) Pub Date : 2016-07-18 13:44:38
Yun Shi, Yong Yang, Yong-Wang Li, Haijun Jiao
Applied Catalysis A: General ( IF 4.7 ) Pub Date : 2016-07-18 13:44:38
Yun Shi, Yong Yang, Yong-Wang Li, Haijun Jiao
|
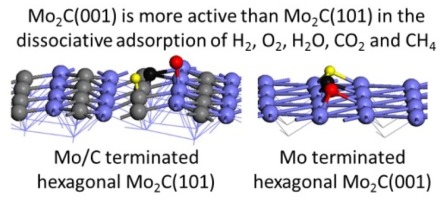
基于自旋极化的周期性密度泛函理论,包括最新的色散校正(PBE-D3),H 2,O 2,H 2 O,CO 2,CO,CH 4和C 2 H x的解离吸附机理计算出六角形的Mo 2 C表面。在我们的研究中,我们使用金属Mo 2 C(001)表面以及Mo / C = 1/1比率的Mo 2 C(101)表面。发现这些小分子的解离吸附是放热的,并且具有低的势垒。并且Mo 2 C(001)表面具有比Mo 2强得多的解离吸附C(101)表面。与Mo 2 C(001)表面相反,OH + H和O + 2H可以在Mo 2 C(101)表面上形成平衡。对于Mo 2 C(001)表面上的C 2 H x解离吸附,CH键解离在动力学上比CC键解离更有利,并且最佳C 2 H 6解离路径遵循C 2 H 6 →CH的顺序。3 CH 2 + H→C 2 H 4 + 2H→CH 2 CH + 3H→C 2 H 2 + 4H→C 2 H + 5H→C 2+6小时。由于非常强的离解吸附能,使用H 2 O可以很容易地氧化两个表面。并且可以预期到高的氧气覆盖率。Mo 2 C(001)表面比Mo 2 C(101)表面可以吸收更多的表面O原子。这些表面也可以通过使用CH 4渗碳,尽管程度不大。与CO 2 + 4H共吸附的表面;与CO 2(CO 2 + H→HCOO或COOH)和CO(CO + H→HCO或COH)的氢化相比,CO 2解离(CO 2 →CO + O→C + 2O)更有利。注意到CO 2加氢成CH 4在Mo 2 C(001)表面上形成的可能性不大,而通过2O和2OH预覆盖的表面可以降低Mo 2 C(101)表面上表面C氢化的有效势垒。

"点击查看英文标题和摘要"
更新日期:2016-07-19
"点击查看英文标题和摘要"
































 京公网安备 11010802027423号
京公网安备 11010802027423号