当前位置:
X-MOL 学术
›
J. Mol. Struct.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Crystal structure and thermodynamic properties of the coordination compound calcium D-gluconate Ca[D-C6H11O7]2(s)
Journal of Molecular Structure ( IF 4.0 ) Pub Date : 2021-02-01 , DOI: 10.1016/j.molstruc.2020.128818 You-Ying Di , Guo-Chun Zhang , Yu-Pu Liu , Yu-Xia Kong , Chun-Sheng Zhou
Journal of Molecular Structure ( IF 4.0 ) Pub Date : 2021-02-01 , DOI: 10.1016/j.molstruc.2020.128818 You-Ying Di , Guo-Chun Zhang , Yu-Pu Liu , Yu-Xia Kong , Chun-Sheng Zhou
|
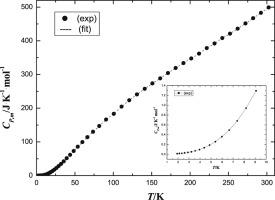
Abstract The coordination compound calcium D-gluconate, Ca[D-C6H11O7]2(s), was synthesized and characterized by chemical analysis, elemental analysis, and X-ray crystallography. Single crystal X-ray diffraction technique revealed that the compound was formed by two D-gluconate anions and one calcium (II) cation. And the D-gluconate anion had a curved chain configuration with an intramolecular bond. The compound exhibited an outstanding chelate property of D-gluconate anions to calcium (II) cations, and the calcium (II) cation was eight-coordinated and chelated by four D-gluconate anions. The lattice potential energy and ionic volume of the anion were calculated to be 1434.05 kJ⋅mol−1 and 0.4211 nm3 from crystallographic data. In accordance with famous Hess law, a reasonable thermochemical cycle was designed and the standard molar enthalpy of formation of Ca[D-C6H11O7]2(s) was calculated as Δ s H m [Ca[D-C6H11O7]2, s] = -(3545.19 ± 1.07) kJ⋅mol−1 by use of an isoperibol solution-reaction calorimeter. Furthermore, molar heat capacities of the compound were measured using a Quantum Design Physical Properties Measurement System (PPMS) with specific heat option within the temperature range from (1.9–300) K. The heat capacities of the compound increased with the temperature and no thermal anomaly was found in the whole temperature region. The experimental data was fitted to a function of the absolute temperature T with a series of theoretical and empirical models for the proper temperature ranges. The values of standard thermodynamic function, C p , m o /J⋅K−1⋅mol−1, Δ 0 T H m o /kJ⋅mol−1, Δ 0 T S m o /J⋅K−1⋅mol−1, and Δ o T G m o / T /J⋅K−1⋅mol−1 (= Δ 0 T S m o - Δ 0 T H m o /T) from T = (0–300) K was calculated based on the fitting results. The standard molar heat capacity, entropy and enthalpy of the compound at T = 298.15 K and 0.1 MPa was determined to be C p , m o = (493.20 ± 2.70) J·K−1 mol−1, H m o = (75934 ± 805) J·mol−1, S m o = (471.55 ± 2.78) J·K−1 mol−1, and G m o / T = - (64658 ± 808) J·K−1⋅mol−1, respectively.
中文翻译:

配位化合物D-葡萄糖酸钙Ca[D-C6H11O7]2(s)的晶体结构和热力学性质
摘要 合成了配位化合物D-葡萄糖酸钙Ca[D-C6H11O7]2(s),并通过化学分析、元素分析和X射线晶体学表征。单晶 X 射线衍射技术表明该化合物由两个 D-葡萄糖酸阴离子和一个钙 (II) 阳离子形成。D-葡萄糖酸阴离子具有带分子内键的弯曲链构型。该化合物表现出优异的D-葡萄糖酸阴离子对钙(II)阳离子的螯合特性,钙(II)阳离子被四个D-葡萄糖酸阴离子螯合八配位。根据晶体学数据计算出阴离子的晶格势能和离子体积为 1434.05 kJ⋅mol−1 和 0.4211 nm3。根据著名的赫斯定律,设计了合理的热化学循环,Ca[D-C6H11O7]2(s) 的标准摩尔生成焓计算为 Δs H m [Ca[D-C6H11O7]2, s] = -(3545.19 ± 1.07) kJ ⋅mol−1 使用 isoperibol 溶液反应量热计。此外,化合物的摩尔热容是使用量子设计物理特性测量系统 (PPMS) 测量的,比热选项在 (1.9-300) K 的温度范围内。化合物的热容随温度增加而没有热整个温区均出现异常。实验数据被拟合为绝对温度 T 的函数,具有一系列适用于适当温度范围的理论和经验模型。标准热力学函数值 C p , mo /J⋅K−1⋅mol−1, Δ 0 TH mo /kJ⋅mol−1, Δ 0 TS mo /J⋅K−1⋅mol−1, 和 Δ o TG mo / T /J⋅K−1⋅mol−1 (= Δ 0 TS mo - Δ 0 TH mo /T) 从 T = ( 0-300) K 是根据拟合结果计算的。化合物在 T = 298.15 K 和 0.1 MPa 下的标准摩尔热容、熵和焓确定为 C p , mo = (493.20 ± 2.70) J·K−1 mol−1, H mo = (75934 ± 805 ) J·mol−1, S mo = (471.55 ± 2.78) J·K−1 mol−1, 和 G mo / T = - (64658 ± 808) J·K−1·mol−1。
更新日期:2021-02-01
中文翻译:
配位化合物D-葡萄糖酸钙Ca[D-C6H11O7]2(s)的晶体结构和热力学性质
摘要 合成了配位化合物D-葡萄糖酸钙Ca[D-C6H11O7]2(s),并通过化学分析、元素分析和X射线晶体学表征。单晶 X 射线衍射技术表明该化合物由两个 D-葡萄糖酸阴离子和一个钙 (II) 阳离子形成。D-葡萄糖酸阴离子具有带分子内键的弯曲链构型。该化合物表现出优异的D-葡萄糖酸阴离子对钙(II)阳离子的螯合特性,钙(II)阳离子被四个D-葡萄糖酸阴离子螯合八配位。根据晶体学数据计算出阴离子的晶格势能和离子体积为 1434.05 kJ⋅mol−1 和 0.4211 nm3。根据著名的赫斯定律,设计了合理的热化学循环,Ca[D-C6H11O7]2(s) 的标准摩尔生成焓计算为 Δs H m [Ca[D-C6H11O7]2, s] = -(3545.19 ± 1.07) kJ ⋅mol−1 使用 isoperibol 溶液反应量热计。此外,化合物的摩尔热容是使用量子设计物理特性测量系统 (PPMS) 测量的,比热选项在 (1.9-300) K 的温度范围内。化合物的热容随温度增加而没有热整个温区均出现异常。实验数据被拟合为绝对温度 T 的函数,具有一系列适用于适当温度范围的理论和经验模型。标准热力学函数值 C p , mo /J⋅K−1⋅mol−1, Δ 0 TH mo /kJ⋅mol−1, Δ 0 TS mo /J⋅K−1⋅mol−1, 和 Δ o TG mo / T /J⋅K−1⋅mol−1 (= Δ 0 TS mo - Δ 0 TH mo /T) 从 T = ( 0-300) K 是根据拟合结果计算的。化合物在 T = 298.15 K 和 0.1 MPa 下的标准摩尔热容、熵和焓确定为 C p , mo = (493.20 ± 2.70) J·K−1 mol−1, H mo = (75934 ± 805 ) J·mol−1, S mo = (471.55 ± 2.78) J·K−1 mol−1, 和 G mo / T = - (64658 ± 808) J·K−1·mol−1。



















































 京公网安备 11010802027423号
京公网安备 11010802027423号