当前位置:
X-MOL 学术
›
Appl. Surf. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Surface Property of the Cu Doped γ-Al2O3:A Density Functional Theory Study
Applied Surface Science ( IF 6.3 ) Pub Date : 2021-01-01 , DOI: 10.1016/j.apsusc.2020.147651 Liu Shi , Yan Huang , Zhang-Hui Lu , Wanglai Cen , Xiaohu Yu , Shaojun Qing , Zhixian Gao , Rongbin Zhang , Gang Feng
Applied Surface Science ( IF 6.3 ) Pub Date : 2021-01-01 , DOI: 10.1016/j.apsusc.2020.147651 Liu Shi , Yan Huang , Zhang-Hui Lu , Wanglai Cen , Xiaohu Yu , Shaojun Qing , Zhixian Gao , Rongbin Zhang , Gang Feng
|
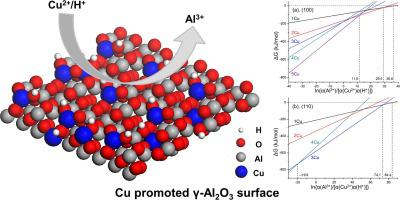
Abstract Cu/γ-Al2O3 catalysts are widely used in many catalytic processes. Investigation into the catalysts structure at molecular level is the basis for the elucidation of the reaction mechanisms and favors the developments of the catalysts. In the present work, periodic density functional theory calculations were performed to investigate the interface of alumina with copper oxides. The interface model is chosen as the substitution of the surface Al atoms of γ-Al2O3 with Cu, and H is used as the ion for charge balance. It is found that the substitution of surface Al3+ by Cu2+ is thermodynamically accessible. Gibbs free energy calculations show that the dehydration temperature for the γ-Al2O3 (1 1 0) surface after substitution is higher than that of on the original γ-Al2O3 (1 1 0) and CuAl2O4 surface. The oxygen vacancy formation energies for the (1 0 0)-5Cu-dehy-2w and (1 1 0)-4Cu-dehy-2w are 213 and 367 kJ/mol, respectively. In addition, the Cu doped γ-Al2O3 interface could strengthen the binding of Cu with the alumina surface. The results provide molecular level insights for the understanding of the interface structures and physical chemistry properties of alumina with copper oxides.
中文翻译:

Cu掺杂γ-Al2O3的表面特性:密度泛函理论研究
摘要 Cu/γ-Al2O3 催化剂广泛应用于许多催化过程。在分子水平上研究催化剂结构是阐明反应机理的基础,有利于催化剂的发展。在目前的工作中,进行了周期性密度泛函理论计算以研究氧化铝与氧化铜的界面。界面模型选用Cu取代γ-Al2O3表面Al原子,H作为电荷平衡离子。发现表面Al3+ 被Cu2+ 取代是热力学上可接近的。Gibbs自由能计算表明,取代后γ-Al2O3(1 1 0)表面的脱水温度高于原始γ-Al2O3(1 1 0)和CuAl2O4表面的脱水温度。(1 0 0)-5Cu-dehy-2w和(1 1 0)-4Cu-dehy-2w的氧空位形成能分别为213和367 kJ/mol。此外,Cu掺杂的γ-Al2O3界面可以加强Cu与氧化铝表面的结合。结果为理解氧化铝与氧化铜的界面结构和物理化学性质提供了分子水平的见解。
更新日期:2021-01-01
中文翻译:
Cu掺杂γ-Al2O3的表面特性:密度泛函理论研究
摘要 Cu/γ-Al2O3 催化剂广泛应用于许多催化过程。在分子水平上研究催化剂结构是阐明反应机理的基础,有利于催化剂的发展。在目前的工作中,进行了周期性密度泛函理论计算以研究氧化铝与氧化铜的界面。界面模型选用Cu取代γ-Al2O3表面Al原子,H作为电荷平衡离子。发现表面Al3+ 被Cu2+ 取代是热力学上可接近的。Gibbs自由能计算表明,取代后γ-Al2O3(1 1 0)表面的脱水温度高于原始γ-Al2O3(1 1 0)和CuAl2O4表面的脱水温度。(1 0 0)-5Cu-dehy-2w和(1 1 0)-4Cu-dehy-2w的氧空位形成能分别为213和367 kJ/mol。此外,Cu掺杂的γ-Al2O3界面可以加强Cu与氧化铝表面的结合。结果为理解氧化铝与氧化铜的界面结构和物理化学性质提供了分子水平的见解。





























 京公网安备 11010802027423号
京公网安备 11010802027423号