当前位置:
X-MOL 学术
›
WIREs Comput. Mol. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Atomistic modeling of electrocatalysis: Are we there yet?
Wiley Interdisciplinary Reviews: Computational Molecular Science ( IF 16.8 ) Pub Date : 2020-08-26 , DOI: 10.1002/wcms.1499 Nawras Abidi 1 , Kang Rui Garrick Lim 2 , Zhi Wei Seh 2 , Stephan N. Steinmann 1
Wiley Interdisciplinary Reviews: Computational Molecular Science ( IF 16.8 ) Pub Date : 2020-08-26 , DOI: 10.1002/wcms.1499 Nawras Abidi 1 , Kang Rui Garrick Lim 2 , Zhi Wei Seh 2 , Stephan N. Steinmann 1
Affiliation
|
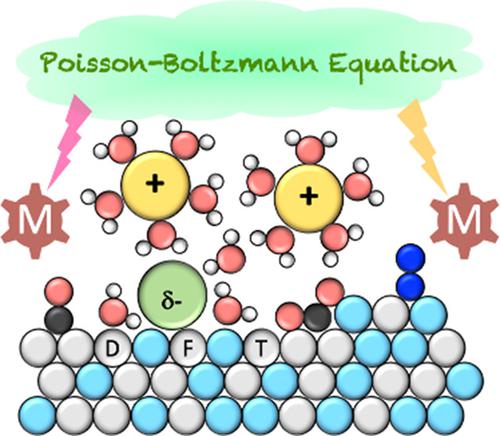
Electrified interfaces play a prime role in energy technologies, from batteries and capacitors to heterogeneous electrocatalysis. The atomistic understanding and modeling of these interfaces is challenging due to the structural complexity and the presence of the electrochemical potential. Including the potential explicitly in the quantum mechanical simulations is equivalent to simulating systems with a surface charge. For realistic relationships between the potential and the surface charge (i.e., the capacity), the solvent and counter charge need to be considered. The solvent and electrolyte description are limited by the computational power: either molecules or ions are included explicitly or implicit solvent and electrolyte descriptions are adopted. The first option is limited by the phase‐space sampling that is at least 10 times too small to reach convergence, while the second is missing a realistic structuring of the interface. Both approaches suffer from a lack of validation against directly comparable experimental data. Furthermore, the limitations of density functional theory in terms of accuracy are critical for these metal/liquid interfaces. Nevertheless, the atomistic insight in electrocatalytic interfaces allows insights with unprecedented details. The joint theoretical and experimental efforts to design non‐noble hydrogen evolution catalysts are discussed as an example for the success of theory to spur and accelerate experimental discoveries.
中文翻译:

电催化的原子建模:我们到了吗?
电气化接口在能源技术中发挥着主要作用,从电池和电容器到多相电催化。由于结构的复杂性和电化学势的存在,对这些界面的原子性理解和建模具有挑战性。在量子力学模拟中明确包含电势等效于模拟具有表面电荷的系统。对于电势和表面电荷(即容量)之间的现实关系,需要考虑溶剂电荷和反电荷。溶剂和电解质的描述受计算能力的限制:显式包括分子或离子,或者采用隐式溶剂和电解质的描述。第一种选择受到相空间采样的限制,相移采样至少小到无法达到收敛的10倍,而第二种却缺少接口的实际结构。两种方法都缺乏针对直接可比的实验数据的验证。此外,就这些金属/液体界面而言,密度泛函理论在准确性方面的局限性至关重要。尽管如此,电催化界面中的原子洞察力仍可提供前所未有的细节洞察力。讨论了设计非贵族氢气析出催化剂的联合理论和实验工作,以此作为理论成功促进和加速实验发现的一个例子。两种方法都缺乏针对直接可比的实验数据的验证。此外,就这些金属/液体界面而言,密度泛函理论在准确性方面的局限性至关重要。尽管如此,电催化界面中的原子洞察力仍可提供前所未有的细节洞察力。讨论了设计非贵族氢气析出催化剂的联合理论和实验工作,以此作为理论成功促进和加速实验发现的一个例子。两种方法都缺乏针对直接可比的实验数据的验证。此外,就这些金属/液体界面而言,密度泛函理论在准确性方面的局限性至关重要。尽管如此,电催化界面中的原子洞察力仍可提供前所未有的细节洞察力。讨论了设计非贵族氢气析出催化剂的联合理论和实验工作,以此作为理论成功促进和加速实验发现的一个例子。
更新日期:2020-08-26
中文翻译:
电催化的原子建模:我们到了吗?
电气化接口在能源技术中发挥着主要作用,从电池和电容器到多相电催化。由于结构的复杂性和电化学势的存在,对这些界面的原子性理解和建模具有挑战性。在量子力学模拟中明确包含电势等效于模拟具有表面电荷的系统。对于电势和表面电荷(即容量)之间的现实关系,需要考虑溶剂电荷和反电荷。溶剂和电解质的描述受计算能力的限制:显式包括分子或离子,或者采用隐式溶剂和电解质的描述。第一种选择受到相空间采样的限制,相移采样至少小到无法达到收敛的10倍,而第二种却缺少接口的实际结构。两种方法都缺乏针对直接可比的实验数据的验证。此外,就这些金属/液体界面而言,密度泛函理论在准确性方面的局限性至关重要。尽管如此,电催化界面中的原子洞察力仍可提供前所未有的细节洞察力。讨论了设计非贵族氢气析出催化剂的联合理论和实验工作,以此作为理论成功促进和加速实验发现的一个例子。两种方法都缺乏针对直接可比的实验数据的验证。此外,就这些金属/液体界面而言,密度泛函理论在准确性方面的局限性至关重要。尽管如此,电催化界面中的原子洞察力仍可提供前所未有的细节洞察力。讨论了设计非贵族氢气析出催化剂的联合理论和实验工作,以此作为理论成功促进和加速实验发现的一个例子。两种方法都缺乏针对直接可比的实验数据的验证。此外,就这些金属/液体界面而言,密度泛函理论在准确性方面的局限性至关重要。尽管如此,电催化界面中的原子洞察力仍可提供前所未有的细节洞察力。讨论了设计非贵族氢气析出催化剂的联合理论和实验工作,以此作为理论成功促进和加速实验发现的一个例子。






























 京公网安备 11010802027423号
京公网安备 11010802027423号