当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
A 2 3+ B 2 4+ O 7(A = Lu 3+ –La 3+,B = Zr 4+,Hf 4+的Weberite型,烧绿石和缺陷萤石结构的稳定性的第一性原理研究,Sn 4+和Ti 4+)
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2020-08-24 , DOI: 10.1021/acs.jpcc.0c05443
Ushio Matsumoto 1, 2 , Takafumi Ogawa 1 , Satoshi Kitaoka 3 , Hiroki Moriwake 1 , Isao Tanaka 1, 2
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2020-08-24 , DOI: 10.1021/acs.jpcc.0c05443
Ushio Matsumoto 1, 2 , Takafumi Ogawa 1 , Satoshi Kitaoka 3 , Hiroki Moriwake 1 , Isao Tanaka 1, 2
Affiliation
|
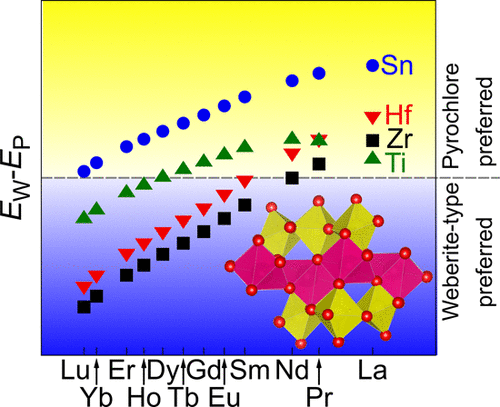
首先,系统地研究了一系列A 2 B 2 O 7(A = Lu 3+ –La 3 +,B = Zr 4+,Hf 4+,Sn 4+和Ti 4+)化合物的晶体结构和能量学。-原理计算。在实验上,已知它们会形成烧绿石或萤石缺陷结构。最近,在Ho 2 Zr 2 O 7中报道了韦伯石型有序的形成。在这项研究中,我们对组成Yb 2 Ti 2进行了详尽的结构搜索O 7,发现最低能量的结构是三斜辉石型结构,其特征在于Yb和Ti层交替。从第一性原理声子计算得出的自由能表明,Yb 2 Ti 2 O 7的韦伯石型和烧绿石结构之间的转变温度为550K 。还比较了韦伯石型和烧绿石结构的能量。A 2 B 2 O 7组成。作为一般趋势,发现随着A 3+的增加,韦伯石型结构的稳定性相对于烧绿石结构增加。阳离子半径减小。还报道了使用特殊的准随机结构估算的无序缺陷萤石结构的能量。发现当A 3+阳离子半径小时,在A 2 Zr 2 O 7和A 2 Hf 2 O 7中的高温下,这种缺陷萤石结构的形成在能量上更有利。目前的结果与先前报道的实验结果一致。

"点击查看英文标题和摘要"
更新日期:2020-09-18
"点击查看英文标题和摘要"
































 京公网安备 11010802027423号
京公网安备 11010802027423号