当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
First Principles Nonadiabatic Excited-State Molecular Dynamics in NWChem.
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-08-18 , DOI: 10.1021/acs.jctc.0c00295 Huajing Song 1 , Sean A Fischer 2 , Yu Zhang 1 , Christopher J Cramer 3 , Shaul Mukamel 4 , Niranjan Govind 5 , Sergei Tretiak 1, 6
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-08-18 , DOI: 10.1021/acs.jctc.0c00295 Huajing Song 1 , Sean A Fischer 2 , Yu Zhang 1 , Christopher J Cramer 3 , Shaul Mukamel 4 , Niranjan Govind 5 , Sergei Tretiak 1, 6
Affiliation
|
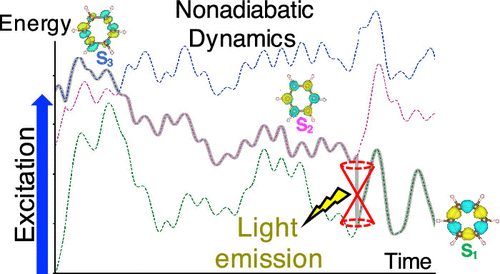
Computational simulation of nonadiabatic molecular dynamics is an indispensable tool for understanding complex photoinduced processes such as internal conversion, energy transfer, charge separation, and spatial localization of excitons, to name a few. We report an implementation of the fewest-switches surface-hopping algorithm in the NWChem computational chemistry program. The surface-hopping method is combined with linear-response time-dependent density functional theory calculations of adiabatic excited-state potential energy surfaces. To treat quantum transitions between arbitrary electronic Born–Oppenheimer states, we have implemented both numerical and analytical differentiation schemes for derivative nonadiabatic couplings. A numerical approach for the time-derivative nonadiabatic couplings together with an analytical method for calculating nonadiabatic coupling vectors is an efficient combination for surface-hopping approaches. Additionally, electronic decoherence schemes and a state reassigned unavoided crossings algorithm are implemented to improve the accuracy of the simulated dynamics and to handle trivial unavoided crossings. We apply our code to study the ultrafast decay of photoexcited benzene, including a detailed analysis of the potential energy surface, population decay timescales, and vibrational coordinates coupled to the excitation dynamics. We also study the photoinduced dynamics in trans-distyrylbenzene. This study provides a baseline for future implementations of higher-level frameworks for simulating nonadiabatic molecular dynamics in NWChem.
中文翻译:

NWChem中的第一原理非绝热激发态分子动力学。
非绝热分子动力学的计算模拟是了解复杂的光诱导过程(例如内部转化,能量转移,电荷分离和激子的空间定位)的必不可少的工具。我们报告了NWChem计算化学程序中最少的开关表面跳跃算法的实现。表面跳变方法与绝热激发态势能面的线性响应时间相关的密度泛函理论计算相结合。为了处理任意电子Born–Oppenheimer态之间的量子跃迁,我们已经为导数非绝热耦合实现了数值和解析微分方案。时间导数非绝热耦合的数值方法以及计算非绝热耦合向量的解析方法是表面跳变方法的有效组合。另外,实施电子去相干方案和状态重新分配的不可避免的穿越算法,以提高模拟动力学的准确性并处理琐碎的不可避免的穿越。我们应用我们的代码研究光激发苯的超快衰减,包括对势能面,人口衰减时标以及与激发动力学耦合的振动坐标的详细分析。我们还研究了反式二苯乙烯基苯中的光诱导动力学。这项研究为在NWChem中模拟非绝热分子动力学的高级框架的未来实现提供了基线。
更新日期:2020-10-13
中文翻译:
NWChem中的第一原理非绝热激发态分子动力学。
非绝热分子动力学的计算模拟是了解复杂的光诱导过程(例如内部转化,能量转移,电荷分离和激子的空间定位)的必不可少的工具。我们报告了NWChem计算化学程序中最少的开关表面跳跃算法的实现。表面跳变方法与绝热激发态势能面的线性响应时间相关的密度泛函理论计算相结合。为了处理任意电子Born–Oppenheimer态之间的量子跃迁,我们已经为导数非绝热耦合实现了数值和解析微分方案。时间导数非绝热耦合的数值方法以及计算非绝热耦合向量的解析方法是表面跳变方法的有效组合。另外,实施电子去相干方案和状态重新分配的不可避免的穿越算法,以提高模拟动力学的准确性并处理琐碎的不可避免的穿越。我们应用我们的代码研究光激发苯的超快衰减,包括对势能面,人口衰减时标以及与激发动力学耦合的振动坐标的详细分析。我们还研究了反式二苯乙烯基苯中的光诱导动力学。这项研究为在NWChem中模拟非绝热分子动力学的高级框架的未来实现提供了基线。
































 京公网安备 11010802027423号
京公网安备 11010802027423号