当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
分子动力学仿真中用于精确力计算的机器学习。
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2020-07-29 , DOI: 10.1021/acs.jpca.0c03926
Punyaslok Pattnaik 1 , Shampa Raghunathan 1 , Tarun Kalluri 2 , Prabhakar Bhimalapuram 1 , C V Jawahar 2 , U Deva Priyakumar 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2020-07-29 , DOI: 10.1021/acs.jpca.0c03926
Punyaslok Pattnaik 1 , Shampa Raghunathan 1 , Tarun Kalluri 2 , Prabhakar Bhimalapuram 1 , C V Jawahar 2 , U Deva Priyakumar 1
Affiliation
|
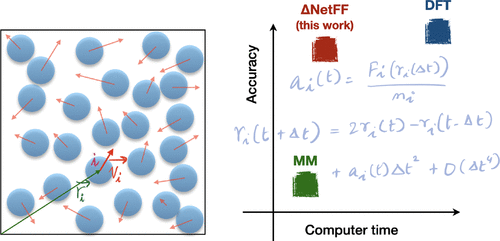
的计算量很大性质从头分子动力学模拟严重限制了其模拟大型系统规模和长时间规模的能力,这两者都是模仿实验条件所必需的。在这项工作中,我们探索一种在小型系统上利用通过量子力学密度泛函理论(DFT)获得的数据的方法,并通过使用深度学习以液氩作为测试案例来随后模拟大型系统。选择合适的矢量表示法来表示每个Ar原子的周围环境,并引入了Δ-NetFF机器学习模型,在该模型中训练了神经网络以预测DFT和经典力场获得的合力的差异。然后根据我们计算的特性,使用来自神经网络的力对各种系统大小和时标执行分子动力学模拟。从经典力场和神经网络模型获得的性能进行了比较,并提供了可用的实验数据以验证所提出的方法。

"点击查看英文标题和摘要"
更新日期:2020-08-27
"点击查看英文标题和摘要"
































 京公网安备 11010802027423号
京公网安备 11010802027423号