当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
A New Workflow for the Analysis of Phosphosite Occupancy in Paired Samples by Integration of Proteomics and Phosphoproteomics Data Sets.
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2020-07-29 , DOI: 10.1021/acs.jproteome.0c00345
Yan Wang 1, 2 , Yu Tian 3, 4 , Xiaoyan Liu 1 , Jing Dong 1 , Liming Wang 4 , Mingliang Ye 1, 2
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2020-07-29 , DOI: 10.1021/acs.jproteome.0c00345
Yan Wang 1, 2 , Yu Tian 3, 4 , Xiaoyan Liu 1 , Jing Dong 1 , Liming Wang 4 , Mingliang Ye 1, 2
Affiliation
|
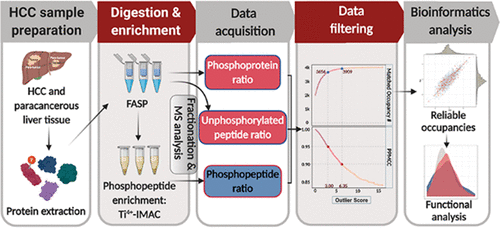
Knowledge of phosphosite occupancy is important to explain biological functions for phosphoproteomics studies. Determination of occupancy using three ratios, i.e., protein ratio, phosphopeptide ratio, and its unmodified peptide counterpart ratio between a pair of samples, is straightforward but suffers from large variances. Here, an optimized protocol of offline fractionation and LC–MS analysis combined with an integrated data processing approach was developed to improve the reliability of the phosphosite occupancy determination. An outlier score S was introduced to evaluate the deviation between the ratio of absolute occupancy and relative occupancy and was further used to define the bounds of a credible interval of absolute occupancy. For a preset product–moment correlation coefficient, the credible interval can be resolved through the S value. Using this strategy, more than 176k unique peptide sequences covering 11k protein groups and 32k phosphosites were identified from one paired hepatocellular carcinoma (HCC) sample and about 3000 reliable phosphosite occupancies were finally determined. By bioinformatics analysis, we characterized the biological properties associated with phosphorylation sites with different quantified occupancies from the paired HCC sample. Data are available via ProteomeXchange with identifier PXD019045.
中文翻译:

通过蛋白质组学和蛋白质组学数据集的整合来分析配对样品中的磷酸盐占用率的新工作流程。
磷酸位点的占有率对于解释磷酸化蛋白质组学研究的生物学功能很重要。使用三个比率(即蛋白质比率,磷酸肽比率及其一对样品之间未修饰的肽对应物比率)确定占用率很简单,但存在较大差异。在这里,开发了一种优化的离线分离和LC-MS分析方案,并结合了集成的数据处理方法,以提高磷矿位测定的可靠性。离群值S引入来评估绝对占用率与相对占用率之间的偏差,并进一步用于定义可信的绝对占用率区间的界限。对于预设的乘积矩相关系数,可以通过S值确定可信区间。使用该策略,从一对配对的肝细胞癌(HCC)样品中鉴定了超过176k的独特肽序列,涵盖11k蛋白组和32k磷酸位点,最终确定了约3000个可靠的磷酸位点占有率。通过生物信息学分析,我们从配对的HCC样品中表征了与磷酸化位点相关的生物学特性,这些位点具有不同的量化占有率。数据可通过ProteomeXchange获得,其标识符为PXD019045。
更新日期:2020-09-05
中文翻译:
通过蛋白质组学和蛋白质组学数据集的整合来分析配对样品中的磷酸盐占用率的新工作流程。
磷酸位点的占有率对于解释磷酸化蛋白质组学研究的生物学功能很重要。使用三个比率(即蛋白质比率,磷酸肽比率及其一对样品之间未修饰的肽对应物比率)确定占用率很简单,但存在较大差异。在这里,开发了一种优化的离线分离和LC-MS分析方案,并结合了集成的数据处理方法,以提高磷矿位测定的可靠性。离群值S引入来评估绝对占用率与相对占用率之间的偏差,并进一步用于定义可信的绝对占用率区间的界限。对于预设的乘积矩相关系数,可以通过S值确定可信区间。使用该策略,从一对配对的肝细胞癌(HCC)样品中鉴定了超过176k的独特肽序列,涵盖11k蛋白组和32k磷酸位点,最终确定了约3000个可靠的磷酸位点占有率。通过生物信息学分析,我们从配对的HCC样品中表征了与磷酸化位点相关的生物学特性,这些位点具有不同的量化占有率。数据可通过ProteomeXchange获得,其标识符为PXD019045。






























 京公网安备 11010802027423号
京公网安备 11010802027423号