Journal of Colloid and Interface Science ( IF 9.4 ) Pub Date : 2020-07-26 , DOI: 10.1016/j.jcis.2020.07.112
Yuyao Zhang 1 , Xiaoying Zhu 1 , Xin Li 2 , Baoliang Chen 1
|
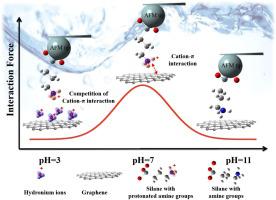
The adsorption of pollutants on carbonaceous environmental media has been widely studied via batch sorption experiments and spectroscopic characterization. However, the molecular interactions between pollutants and interfacial sites on carbonaceous materials have only been indirectly investigated. To comprehend the adsorption mechanisms in situ, we applied atomic force microscopy force spectroscopy (AFM-FS) to quantitatively determine the molecular interactions between typical amines (methylamines and N-methylaniline) and the surface of highly oriented pyrolytic graphite (HOPG), which was supported by the single molecule interaction derived from density functional theory and batch adsorption experiments. This method achieved direct and in situ characterization of the molecular interactions in the adsorption process. The molecular interactions between the amines and the adsorption sites on the graphite surface were affected by pH and peaked at pH 7 due to strong cation-π interactions. When the pH was 11, the attractions were weak due to a lack of cation-π interaction, whereas, when the pH was 3, the competitive occupation of hydronium ions on the surface reduced the attraction between the amines and HOPG. Based on AFM-FS, the single molecule force of methylamine and N-methylaniline on the graphite surface was estimated to be 0.224 nN and 0.153 nN, respectively, which was consistent with density functional theory (DFT) calculations. This study broadens our comprehension of cation-π interactions between amines and electron-rich aromatic compounds at the micro/nanoscale.
中文翻译:
使用原子力显微镜原位定量测定胺和石墨烯表面之间的分子间吸引力。
通过分批吸附实验和光谱表征已广泛研究了污染物在含碳环境介质上的吸附。但是,仅间接研究了污染物与碳质材料上界面部位之间的分子相互作用。为了了解原位吸附机理,我们应用了原子力显微镜力谱(AFM-FS)来定量确定典型胺(甲胺和N-甲基苯胺)与高取向热解石墨(HOPG)表面之间的分子相互作用。由密度泛函理论和分批吸附实验得出的单分子相互作用得到了支持。这种方法实现了直接和原位吸附过程中分子相互作用的表征。胺与石墨表面吸附位点之间的分子相互作用受pH的影响,并且由于强的阳离子-π相互作用而在pH 7达到峰值。pH值为11时,由于缺乏阳离子与π的相互作用,吸引力弱,而pH值为3时,水合氢离子在表面上的竞争性占领降低了胺与HOPG之间的吸引力。基于AFM-FS,甲胺和氮的单分子力石墨表面上的-甲基苯胺估计分别为0.224 nN和0.153 nN,这与密度泛函理论(DFT)计算是一致的。这项研究拓宽了我们对微观/纳米尺度上胺与富电子芳族化合物之间阳离子-π相互作用的理解。
































 京公网安备 11010802027423号
京公网安备 11010802027423号