当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Validation of Local Hybrid Functionals for Excited States: Structures, Fluorescence, Phosphorescence, and Vibronic Spectra.
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-07-22 , DOI: 10.1021/acs.jctc.0c00520 Robin Grotjahn 1 , Martin Kaupp 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-07-22 , DOI: 10.1021/acs.jctc.0c00520 Robin Grotjahn 1 , Martin Kaupp 1
Affiliation
|
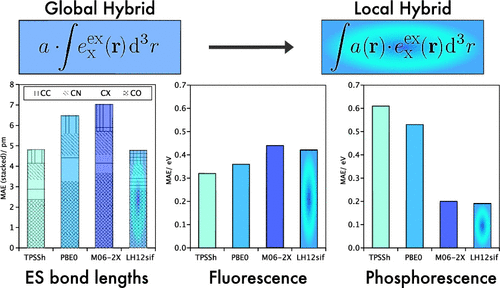
Local hybrid functionals are evaluated in linear-response TDDFT computations for a broad range of excited-state properties including excited-state structures, fluorescence, and phosphorescence energies and the vibronic shape of absorption and phosphorescence spectra. Computation of such properties requires the optimization of excited states, which is facilitated by the recent implementation of excited-state gradients for local hybrid functionals in the TURBOMOLE program (Grotjahn, R.; Furche, F.; Kaupp, M. J. Chem. Theory Comput. 2019, 15, 5508). Comparison with coupled-cluster reference values reveals competitive performance of local hybrids for excited-state bond lengths with particular advantages for carbon–halogen, carbon–carbon, and carbon–nitrogen bonds. As with most global and range-separated hybrid functionals, carbonyl and thionyl bonds in n → π* excited states are found to be too compact. For the emission energies, results depend on the multiplicity of the excited state. While the local hybrid functionals tested perform moderately well, comparable to global hybrids, for singlet states (fluorescence energies), they provide outstanding accuracy for triplet states (phosphorescence energies), only matched by those from the highly empirical M06-2X hybrid functional. The assessment of the shape of vibronic spectra reveals rather small differences between local hybrid functionals and conventional hybrid functionals with comparable exact-exchange admixture. The advantages for phosphorescence energies and the robust performance for the shape of vibronic spectra are combined to showcase the potential of local hybrid functionals for the prediction of phosphorescence spectra.
中文翻译:

验证激发态的局部杂化功能:结构,荧光,磷光和振动光谱。
在线性响应TDDFT计算中评估了局部混合功能,以得到广泛的激发态特性,包括激发态结构,荧光和磷光能以及吸收和磷光光谱的振动波形状。此类性质的计算需要优化激发态,这是由于最近在TURBOMOLE程序中实现了用于局部混合功能的激发态梯度(Grotjahn,R .; Furche,F .; Kaupp,M.J .理论COMPUT。 2019,15,5508)。与耦合簇参考值的比较表明,在激发态键长方面,局部杂化剂具有竞争性,其中碳-卤素键,碳-碳键和碳-氮键具有特殊优势。与大多数全局和范围分隔的杂合官能团一样,n中的羰基和亚硫基键→发现π*激发态太紧凑。对于发射能量,结果取决于激发态的多重性。尽管测试的本地混合功能对于单重态(荧光能量)的性能与全局混合性能相当,但它们为三重态(磷光能量)提供了出色的准确性,仅与高度经验化的M06-2X混合功能相匹配。振动光谱形状的评估表明,在具有可比的精确交换混合物的情况下,本地混合功能与常规混合功能之间的差异很小。结合了磷光能量的优点和对电子频谱形状的鲁棒性能,以展示局部混合功能在预测磷光光谱方面的潜力。
更新日期:2020-09-09
中文翻译:
验证激发态的局部杂化功能:结构,荧光,磷光和振动光谱。
在线性响应TDDFT计算中评估了局部混合功能,以得到广泛的激发态特性,包括激发态结构,荧光和磷光能以及吸收和磷光光谱的振动波形状。此类性质的计算需要优化激发态,这是由于最近在TURBOMOLE程序中实现了用于局部混合功能的激发态梯度(Grotjahn,R .; Furche,F .; Kaupp,M.J .理论COMPUT。 2019,15,5508)。与耦合簇参考值的比较表明,在激发态键长方面,局部杂化剂具有竞争性,其中碳-卤素键,碳-碳键和碳-氮键具有特殊优势。与大多数全局和范围分隔的杂合官能团一样,n中的羰基和亚硫基键→发现π*激发态太紧凑。对于发射能量,结果取决于激发态的多重性。尽管测试的本地混合功能对于单重态(荧光能量)的性能与全局混合性能相当,但它们为三重态(磷光能量)提供了出色的准确性,仅与高度经验化的M06-2X混合功能相匹配。振动光谱形状的评估表明,在具有可比的精确交换混合物的情况下,本地混合功能与常规混合功能之间的差异很小。结合了磷光能量的优点和对电子频谱形状的鲁棒性能,以展示局部混合功能在预测磷光光谱方面的潜力。




















































 京公网安备 11010802027423号
京公网安备 11010802027423号