当前位置:
X-MOL 学术
›
Inorg. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Characterization of the "Absent" Vanadium Oxo V(═O){N(SiMe3)2}3, Imido V(═NSiMe3){N(SiMe3)2}3, and Imido-Siloxy V(═NSiMe3)(OSiMe3){N(SiMe3)2}2 Complexes Derived from V{N(SiMe3)2}3 and Kinetic Study of the Spontaneous Conversion of the Oxo Complex into Its Imido-Siloxy Isomer.
Inorganic Chemistry ( IF 4.3 ) Pub Date : 2020-07-22 , DOI: 10.1021/acs.inorgchem.0c01572 Cary R Stennett 1 , Thien H Nguyen 1 , Philip P Power 1
Inorganic Chemistry ( IF 4.3 ) Pub Date : 2020-07-22 , DOI: 10.1021/acs.inorgchem.0c01572 Cary R Stennett 1 , Thien H Nguyen 1 , Philip P Power 1
Affiliation
|
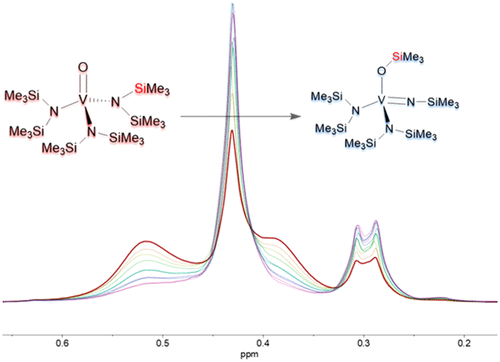
The synthesis and characterization of V(═O){N(SiMe3)2}3 (1), V(═NSiMe3){N(SiMe3)2}3 (2), and V(═NSiMe3)(OSiMe3){N(SiMe3)2}2 (3) are described. Prior attempts to synthesize the vanadium(V) oxo complex 1 via salt metathesis of VOCl3 with the lithium or sodium silylamide salt had yielded either the putative rearranged species V(═NSiMe3)(OSiMe3){N(SiMe3)2}2 (3) or the oxo-bridged, dimetallic {(μ-O)2V2[N(SiMe3)2]4}. We now show that complex 1 is available by treatment of the vanadium(III) tris(silylamide) V{N(SiMe3)2}3 with iodosylbenzene. The imido complex 2 was obtained by treatment of V{N(SiMe3)2}3 with trimethylsilyl azide. Sublimation of 1 formed complex 3, which was determined to be V(═NSiMe3)(OSiMe3){N(SiMe3)2}2, on the basis of infrared, electronic, and 1H and 51V NMR spectroscopies. Crystallographic disorder precluded a complete structural characterization of 3, although a four-coordinate V atom, as well as severely disordered ligands, were apparent. Comparison of the vibrational spectra of 1 and 2 allowed an unambiguous assignment of the V–O (995 cm–1) and V–Nimide (1060 cm–1) stretching bands. The vibrational spectrum of complex 3 displayed strong absorbances at 1090 and 945 cm–1, indicative of its metal imide and metal siloxide moieties. The 1H NMR spectrum of 1 in deuterated benzene showed overlapping signals for the ligand protons proximal and distal to the oxo moiety at 0.52 and 0.38 ppm. The 1H NMR spectrum of 2 in deuterated methylene chloride displayed distinct signals for the imido (0.41 ppm) and amido (0.35 ppm) protons, whereas 1H NMR spectroscopy of 3 showed three signals in an intensity ratio consistent with the formula V(═NSiMe3)(OSiMe3){N(SiMe3)2}2. 51V NMR spectra of 1–3 revealed singlet resonances at −119 ppm (1), −24 ppm (2), and −279 ppm (3). The electronic spectra of 1–3 displayed single absorbances in the charge transfer region, consistent with their d0 electron configurations. Kinetic studies of the spontaneous conversion of complex 1 to 3 were used to determine the rate constants (ca. 0.0002 s–1 (63 °C), 0.0006 s–1 (73 °C), 0.002 s–1 (83 °C)) and activation energy (ca. 20 kcal/mol) of this first-order process.
中文翻译:

“不存在”钒氧V(= O){N(SiMe3)2} 3,亚氨基V(= NSiMe3){N(SiMe3)2} 3和亚氨基-甲硅烷氧基V(═NSiMe3)(OSiMe3){ V {N(SiMe3)2} 3衍生的N(SiMe3)2} 2配合物和羰基配合物自发转化为亚氨基-甲硅烷氧基异构体的动力学研究。
V(= O){N(森达的合成和表征3)2 } 3(1),V(═NSiMe 3){N(森达3)2 } 3(2),和V(═NSiMe 3)(描述了OSiMe 3){N(SiMe 3)2 } 2(3)。之前的尝试来合成氧络合物的钒(V)1经由VOCl的盐复分解3与锂甲硅烷胺或钠盐或者已经产生了推定的重排的物种V(═NSiMe 3)(OSiMe 3){N(森达3)2 } 2(3)或氧桥双金属{(μ-O)2 V 2 [N(SiMe 3)2 ] 4 }。我们现在显示配合物1可通过用碘代基苯处理钒(III)三(甲硅烷基酰胺)V {N(SiMe 3)2 } 3获得。通过用三甲基甲硅烷基叠氮化物处理V {N(SiMe 3)2 } 3来获得亚氨基配合物2。的升华1形成的复合3,其被确定为V(═NSiMe 3)(OSiMe 3){N(SiMe 3)2 } 2,基于红外,电子以及1 H和51 V NMR光谱学。晶体学障碍排除了3的完整结构特征,尽管很明显有四个配位的V原子以及严重无序的配体。通过比较1和2的振动谱,可以明确指定V–O(995 cm –1)和V–N酰亚胺(1060 cm –1)的拉伸带。配合物3的振动光谱在1090和945 cm处显示出强吸收–1,表示其金属酰亚胺和金属氧化硅部分。氘代苯中1的1 H NMR谱图显示在oxo部分的近端和远端,配体质子在0.52和0.38 ppm处有重叠信号。的1的1 H NMR光谱2在氘代二氯甲烷显示不同的信号为酰亚胺(0.41 PPM)和酰氨基(0.35 ppm的)质子,而1个的1 H NMR光谱法3中的强度比与式V(一致表明三个信号═ NSiMe 3)(OSiMe 3){N(SiMe 3)2 } 2。的51 V NMR光谱1 - 3在-119 ppm的(显示单峰共振1),-24 ppm的(2),和-279 ppm的(3)。的电子光谱1 - 3显示在电荷转移区域的单吸光度,用他们d一致0电子配置。对复合物1至3自发转化的动力学研究用于确定速率常数(约0.0002 s –1(63°C),0.0006 s –1(73°C),0.002 s –1(83°C) )和该一级过程的活化能(约20 kcal / mol)。
更新日期:2020-08-03
中文翻译:
“不存在”钒氧V(= O){N(SiMe3)2} 3,亚氨基V(= NSiMe3){N(SiMe3)2} 3和亚氨基-甲硅烷氧基V(═NSiMe3)(OSiMe3){ V {N(SiMe3)2} 3衍生的N(SiMe3)2} 2配合物和羰基配合物自发转化为亚氨基-甲硅烷氧基异构体的动力学研究。
V(= O){N(森达的合成和表征3)2 } 3(1),V(═NSiMe 3){N(森达3)2 } 3(2),和V(═NSiMe 3)(描述了OSiMe 3){N(SiMe 3)2 } 2(3)。之前的尝试来合成氧络合物的钒(V)1经由VOCl的盐复分解3与锂甲硅烷胺或钠盐或者已经产生了推定的重排的物种V(═NSiMe 3)(OSiMe 3){N(森达3)2 } 2(3)或氧桥双金属{(μ-O)2 V 2 [N(SiMe 3)2 ] 4 }。我们现在显示配合物1可通过用碘代基苯处理钒(III)三(甲硅烷基酰胺)V {N(SiMe 3)2 } 3获得。通过用三甲基甲硅烷基叠氮化物处理V {N(SiMe 3)2 } 3来获得亚氨基配合物2。的升华1形成的复合3,其被确定为V(═NSiMe 3)(OSiMe 3){N(SiMe 3)2 } 2,基于红外,电子以及1 H和51 V NMR光谱学。晶体学障碍排除了3的完整结构特征,尽管很明显有四个配位的V原子以及严重无序的配体。通过比较1和2的振动谱,可以明确指定V–O(995 cm –1)和V–N酰亚胺(1060 cm –1)的拉伸带。配合物3的振动光谱在1090和945 cm处显示出强吸收–1,表示其金属酰亚胺和金属氧化硅部分。氘代苯中1的1 H NMR谱图显示在oxo部分的近端和远端,配体质子在0.52和0.38 ppm处有重叠信号。的1的1 H NMR光谱2在氘代二氯甲烷显示不同的信号为酰亚胺(0.41 PPM)和酰氨基(0.35 ppm的)质子,而1个的1 H NMR光谱法3中的强度比与式V(一致表明三个信号═ NSiMe 3)(OSiMe 3){N(SiMe 3)2 } 2。的51 V NMR光谱1 - 3在-119 ppm的(显示单峰共振1),-24 ppm的(2),和-279 ppm的(3)。的电子光谱1 - 3显示在电荷转移区域的单吸光度,用他们d一致0电子配置。对复合物1至3自发转化的动力学研究用于确定速率常数(约0.0002 s –1(63°C),0.0006 s –1(73°C),0.002 s –1(83°C) )和该一级过程的活化能(约20 kcal / mol)。






























 京公网安备 11010802027423号
京公网安备 11010802027423号