当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Ligand Gaussian Accelerated Molecular Dynamics (LiGaMD): Characterization of Ligand Binding Thermodynamics and Kinetics.
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-07-21 , DOI: 10.1021/acs.jctc.0c00395 Yinglong Miao 1 , Apurba Bhattarai 1 , Jinan Wang 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-07-21 , DOI: 10.1021/acs.jctc.0c00395 Yinglong Miao 1 , Apurba Bhattarai 1 , Jinan Wang 1
Affiliation
|
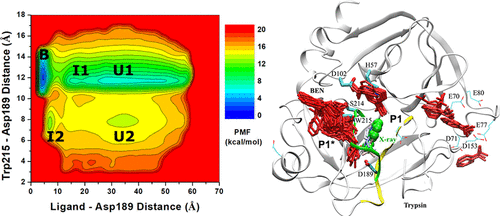
Calculations of ligand binding free energies and kinetic rates are important for drug design. However, such tasks have proven challenging in computational chemistry and biophysics. To address this challenge, we have developed a new computational method, ligand Gaussian accelerated molecular dynamics (LiGaMD), which selectively boosts the ligand nonbonded interaction potential energy based on the Gaussian accelerated molecular dynamics (GaMD) enhanced sampling technique. Another boost potential could be applied to the remaining potential energy of the entire system in a dual-boost algorithm (LiGaMD_Dual) to facilitate ligand binding. LiGaMD has been demonstrated on host–guest and protein–ligand binding model systems. Repetitive guest binding and unbinding in the β-cyclodextrin host were observed in hundreds-of-nanosecond LiGaMD_Dual simulations. The calculated guest binding free energies agreed excellently with experimental data with <1.0 kcal/mol errors. Compared with converged microsecond-time scale conventional molecular dynamics simulations, the sampling errors of LiGaMD_Dual simulations were also <1.0 kcal/mol. Accelerations of ligand kinetic rate constants in LiGaMD simulations were properly estimated using Kramers’ rate theory. Furthermore, LiGaMD allowed us to capture repetitive dissociation and binding of the benzamidine inhibitor in trypsin within 1 μs simulations. The calculated ligand binding free energy and kinetic rate constants compared well with the experimental data. In summary, LiGaMD provides a powerful enhanced sampling approach for characterizing ligand binding thermodynamics and kinetics simultaneously, which is expected to facilitate computer-aided drug design.
中文翻译:

配体高斯加速分子动力学 (LiGaMD):配体结合热力学和动力学的表征。
配体结合自由能和动力学速率的计算对于药物设计很重要。然而,事实证明,这些任务在计算化学和生物物理学中具有挑战性。为了应对这一挑战,我们开发了一种新的计算方法,配体高斯加速分子动力学(LiGaMD),它基于高斯加速分子动力学(GaMD)增强采样技术选择性地提高配体非键相互作用势能。在双增强算法 (LiGaMD_Dual) 中,可以将另一个增强势应用于整个系统的剩余势能,以促进配体结合。LiGaMD 已在宿主 - 客体和蛋白质 - 配体结合模型系统上得到证明。在数百纳秒的 LiGaMD_Dual 模拟中观察到了 β-环糊精宿主中重复的客体结合和解除结合。计算的客体结合自由能与实验数据非常吻合,误差 <1.0 kcal/mol。与收敛的微秒级常规分子动力学模拟相比,LiGaMD_Dual 模拟的采样误差也小于 1.0 kcal/mol。LiGaMD 模拟中配体动力学速率常数的加速度是使用 Kramers 的速率理论正确估计的。此外,LiGaMD 使我们能够在 1 μs 模拟内捕获苯甲脒抑制剂在胰蛋白酶中的重复解离和结合。计算的配体结合自由能和动力学速率常数与实验数据相比较。总之,LiGaMD 提供了一种强大的增强采样方法,可同时表征配体结合热力学和动力学,
更新日期:2020-09-09
中文翻译:
配体高斯加速分子动力学 (LiGaMD):配体结合热力学和动力学的表征。
配体结合自由能和动力学速率的计算对于药物设计很重要。然而,事实证明,这些任务在计算化学和生物物理学中具有挑战性。为了应对这一挑战,我们开发了一种新的计算方法,配体高斯加速分子动力学(LiGaMD),它基于高斯加速分子动力学(GaMD)增强采样技术选择性地提高配体非键相互作用势能。在双增强算法 (LiGaMD_Dual) 中,可以将另一个增强势应用于整个系统的剩余势能,以促进配体结合。LiGaMD 已在宿主 - 客体和蛋白质 - 配体结合模型系统上得到证明。在数百纳秒的 LiGaMD_Dual 模拟中观察到了 β-环糊精宿主中重复的客体结合和解除结合。计算的客体结合自由能与实验数据非常吻合,误差 <1.0 kcal/mol。与收敛的微秒级常规分子动力学模拟相比,LiGaMD_Dual 模拟的采样误差也小于 1.0 kcal/mol。LiGaMD 模拟中配体动力学速率常数的加速度是使用 Kramers 的速率理论正确估计的。此外,LiGaMD 使我们能够在 1 μs 模拟内捕获苯甲脒抑制剂在胰蛋白酶中的重复解离和结合。计算的配体结合自由能和动力学速率常数与实验数据相比较。总之,LiGaMD 提供了一种强大的增强采样方法,可同时表征配体结合热力学和动力学,






























 京公网安备 11010802027423号
京公网安备 11010802027423号