Bioorganic Chemistry ( IF 4.5 ) Pub Date : 2020-07-14 , DOI: 10.1016/j.bioorg.2020.104092 Ruifeng Wang 1 , Xiangxin Zhao 1 , Sijia Yu 1 , Yixuan Chen 2 , Hengxian Cui 1 , Tianxiao Wu 1 , Chenzhou Hao 1 , Dongmei Zhao 1 , Maosheng Cheng 1
|
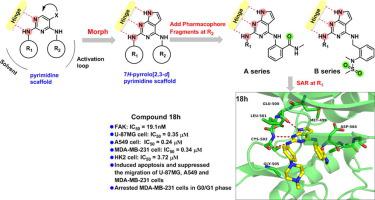
Focal adhesion kinase (FAK) is an intracellular non-receptor tyrosine kinase responsible for development of various tumor types. Aiming to explore new potent inhibitors, two series of 2,4-disubstituted-7H-pyrrolo[2,3-d]pyrimidine derivatives were designed and synthesized on the base of structure-based design strategy. Biological evaluation indicated that most of these new compounds could potently inhibit FAK kinase, leading to the promising inhibitors against the proliferation of U-87MG, A-549, and MDA-MB-231 cancer cell lines. Among them, the optimized compound 18h potently inhibited the enzyme (IC50 = 19.1 nM) and displayed stronger potency than TAE-226 in U-87MG, A-549 and MDA-MB-231 cells, with IC50 values of 0.35, 0.24, and 0.34 μM, respectively. Compound 18h is a multi-target kinase inhibitor. Furthermore, compound 18h also exhibited relatively less cytotoxicity (IC50 = 3.72 μM) toward a normal human cell line, HK2. According to the flow cytometry and wound healing assay results, compound 18h effectively induced apoptosis and G0/G1 phase arrest of MDA-MB-231 cells and suppressed the migration of U-87MG, A-549 and MDA-MB-231 cells. The docking study of compound 18h was performed to elucidate its possible binding modes and to provide a structural basis for the further structural guidance design of FAK inhibitors. Collectively, these data support the further development of compound 18h as a lead compound for FAK-targeted anticancer drug discovery.
中文翻译:
7H-吡咯并[2,3-d]吡啶衍生物作为有效的FAK抑制剂的发现:设计,合成,生物学评估和分子对接研究。
黏着斑激酶(FAK)是负责各种肿瘤类型发展的细胞内非受体酪氨酸激酶。为了探索新的有效抑制剂,在基于结构的设计策略的基础上,设计并合成了两个系列的2,4-二取代-7 H-吡咯并[2,3- d ]嘧啶衍生物。生物学评估表明,这些新化合物中的大多数可以有效抑制FAK激酶,从而导致有希望的抑制剂对抗U-87MG,A-549和MDA-MB-231癌细胞的增殖。其中,优化的化合物18h 在U-87MG,A-549和MDA-MB-231细胞中均有效抑制酶(IC 50 = 19.1 nM),并显示出比TAE-226更强的效力,IC 50分别为0.35、0.24和0.34μM。化合物18h是多靶激酶抑制剂。此外,化合物18h 对正常人细胞株HK2的细胞毒性也较小(IC 50 = 3.72μM)。根据流式细胞术和伤口愈合试验结果,化合物18h有效诱导MDA-MB-231细胞凋亡和G0 / G1期阻滞,并抑制U-87MG,A-549和MDA-MB-231细胞的迁移。进行化合物18h的对接研究以阐明其可能的结合方式,并为FAK抑制剂的进一步结构指导设计提供结构基础。这些数据共同支持化合物18h的进一步开发 作为FAK靶向抗癌药物发现的先导化合物。





























 京公网安备 11010802027423号
京公网安备 11010802027423号