当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
DFT-Based Cu(111)||Cu2O(111) Model for Copper Metal Covered by Ultrathin Copper Oxide: Structure, Electronic Properties, and Reactivity
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2020-07-09 , DOI: 10.1021/acs.jpcc.0c04453
Fatah Chiter 1 , Dominique Costa 1 , Vincent Maurice 1 , Philippe Marcus 1
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2020-07-09 , DOI: 10.1021/acs.jpcc.0c04453
Fatah Chiter 1 , Dominique Costa 1 , Vincent Maurice 1 , Philippe Marcus 1
Affiliation
|
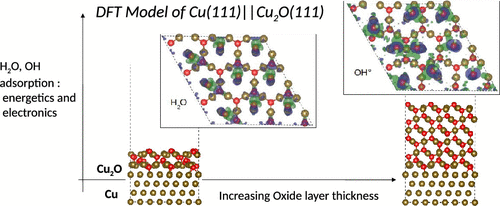
In many applications, metallic copper can be covered by an ultrathin native or a passive oxide film depending on the redox conditions of the interface formed with the environment, which governs its surface properties. In the present work, we constructed and optimized through quantum chemical density functional theory (DFT) calculations a model of this Cu(111)||Cu2O(111) stoichiometric oxide supported in parallel epitaxy on a Cu(111) substrate. Energetic analysis of the metal–oxide interface shows the stable formation of the interface between the oxide film and metal substrate. The Cu2O(111) film binds to the Cu(111) surface via the unsaturated oxygen atoms bonded to copper metal atoms in top, bridge, and hollow sites. Structural analysis of the Cu2O(111) film surface shows reconstruction with modifications of the distances between the unsaturated and the saturated copper atoms. The surface interlayer relaxation is inward for Cu(111)||Cu2O(111), whereas it is outward for the nonsupported Cu2O(111) oxide and converges to that of bulk oxide with increasing the Cu2O(111) thickness. The electronic work function is not significantly modified with respect to the Cu(111) electronic work function, whatever the oxide thickness. The hydration of the surface is energetically favorable, and water molecular adsorption is favored over dissociative adsorption. Water molecules chemisorb on top over unsaturated copper atoms. The hydroxylation of the surface shows two adsorption modes of the OH groups on top over unsaturated Cu atoms and on a bridge site between unsaturated and saturated Cu atoms.
中文翻译:

超薄氧化铜覆盖的铜金属的基于DFT的Cu(111)|| Cu 2 O(111)模型:结构,电子性能和反应性
在许多应用中,取决于与环境形成的界面的氧化还原条件,金属铜可以被超薄的天然或被动氧化膜覆盖,从而决定了其表面性能。在目前的工作中,我们通过量子化学密度泛函理论(DFT)计算构建并优化了平行外延负载在Cu(111)衬底上的Cu(111)|| Cu 2 O(111)化学计量氧化物的模型。对金属-氧化物界面的能量分析表明,氧化膜和金属基底之间界面的稳定形成。Cu 2 O(111)膜通过不饱和氧原子键合到Cu(111)表面,该不饱和氧原子键合在顶部,桥臂和中空部位的铜金属原子上。Cu 2的结构分析O(111)膜表面显示出不饱和和饱和铜原子之间的距离发生了变化的重构。Cu(111)|| Cu 2 O(111)的表面层间弛豫向内,而无支撑的Cu 2 O(111)氧化物的表面层间弛豫向外,并随着Cu 2的增加而收敛于块状氧化物。O(111)厚度。不论氧化物厚度如何,相对于Cu(111)电子功函数,电子功函数均未得到明显修改。表面的水合在能量上是有利的,并且水分子吸附优于解离吸附。水分子在不饱和铜原子上方化学吸附。表面的羟基化显示在不饱和Cu原子上方和在不饱和Cu原子与饱和Cu原子之间的桥键位上的OH基有两种吸附模式。
更新日期:2020-08-06
中文翻译:
超薄氧化铜覆盖的铜金属的基于DFT的Cu(111)|| Cu 2 O(111)模型:结构,电子性能和反应性
在许多应用中,取决于与环境形成的界面的氧化还原条件,金属铜可以被超薄的天然或被动氧化膜覆盖,从而决定了其表面性能。在目前的工作中,我们通过量子化学密度泛函理论(DFT)计算构建并优化了平行外延负载在Cu(111)衬底上的Cu(111)|| Cu 2 O(111)化学计量氧化物的模型。对金属-氧化物界面的能量分析表明,氧化膜和金属基底之间界面的稳定形成。Cu 2 O(111)膜通过不饱和氧原子键合到Cu(111)表面,该不饱和氧原子键合在顶部,桥臂和中空部位的铜金属原子上。Cu 2的结构分析O(111)膜表面显示出不饱和和饱和铜原子之间的距离发生了变化的重构。Cu(111)|| Cu 2 O(111)的表面层间弛豫向内,而无支撑的Cu 2 O(111)氧化物的表面层间弛豫向外,并随着Cu 2的增加而收敛于块状氧化物。O(111)厚度。不论氧化物厚度如何,相对于Cu(111)电子功函数,电子功函数均未得到明显修改。表面的水合在能量上是有利的,并且水分子吸附优于解离吸附。水分子在不饱和铜原子上方化学吸附。表面的羟基化显示在不饱和Cu原子上方和在不饱和Cu原子与饱和Cu原子之间的桥键位上的OH基有两种吸附模式。
































 京公网安备 11010802027423号
京公网安备 11010802027423号