当前位置:
X-MOL 学术
›
CrystEngComm
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Molecular simulation studies on the design of energetic ammonium dinitramide co-crystals for tuning hygroscopicity
CrystEngComm ( IF 2.6 ) Pub Date : 2020-07-09 , DOI: 10.1039/d0ce00602e Zhongqi Ren 1, 2, 3, 4 , Xinjian Chen 1, 2, 3, 4 , Guojia Yu 1, 2, 3, 4 , Yinglei Wang 4, 5, 6 , Bin Chen 4, 5, 6 , Zhiyong Zhou 1, 2, 3, 4
CrystEngComm ( IF 2.6 ) Pub Date : 2020-07-09 , DOI: 10.1039/d0ce00602e Zhongqi Ren 1, 2, 3, 4 , Xinjian Chen 1, 2, 3, 4 , Guojia Yu 1, 2, 3, 4 , Yinglei Wang 4, 5, 6 , Bin Chen 4, 5, 6 , Zhiyong Zhou 1, 2, 3, 4
Affiliation
|
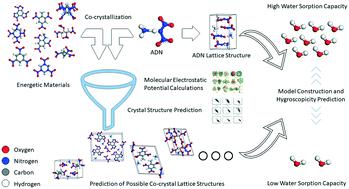
Cocrystallization technology is an effective method for improving crystal properties. Ammonium dinitramide (ADN) is an important component of propellants. However, the high hygroscopicity property of ADN limits its applications. In order to solve this problem, nine energetic co-formers containing 2,4,5,8,10,12-hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane (CL-20), benzotrifuroxan (BTF), 1,3,5,7-tetranitro-1,3,5,7-tetrazocane (HMX), 2,4,6-trinitrotoluene (TNT), butane-1,2,3,4-tetrayl tetranitrate (ETN), hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX), 1,3,5-trinitrobenzene (TNB), 2,4,6-trinitro-N-methylaniline (MATNB), and 1,3,3- trinitroazetidine (TNAZ) were selected to predict the hygroscopicity of co-crystals with the molar ratio of ADN to co-former as 1 : 1. A novel computational method has been put forward to select co-formers efficiently for designing energetic co-crystals with a low water sorption capacity. Based on the molecular electrostatic potential calculations, the intermolecular binding sites in the co-formers of co-crystal were found. By the lattice energy minimization methodology, the possible crystal structures were constructed, and the density and H-bond energy of possible energetic co-crystals were predicted. The co-crystal screening method was used to calculate pairwise interactions and select promising co-crystal formers for the experimental screening. This method can be used to predict the co-crystal hygroscopicity, which thus may speed up the progress of developing novel energetic co-crystal materials.
中文翻译:

调节吸湿性的高能二硝酰胺铵共晶体设计的分子模拟研究
共结晶技术是提高晶体性能的有效方法。二硝酰胺铵(ADN)是推进剂的重要成分。但是,ADN的高吸湿性限制了其应用。为了解决此问题,九种含能的共形成物,其中包含2,4,5,8,10,12-六硝基-2,4,6,8,10,12-六氮杂异纤锌矿型结构烷烃(CL-20),苯并三呋喃(BTF) ,1,3,5,7-四硝基-1,3,5,7-四唑烷(HMX),2,4,6-三硝基甲苯(TNT),丁烷-1,2,3,4-四硝酸四丁酯(ETN) ,六氢-1,3,5-三硝基-1,3,5-三嗪(RDX),1,3,5-三硝基苯(TNB),2,4,6-三硝基-N-甲基苯胺(MATNB)和1选择1,3,3-三硝基氮杂环丁烷(TNAZ)来预测共晶体的吸湿性,其中ADN与共形成物的摩尔比为1:1。提出了一种新颖的计算方法来有效地选择共形成物以设计具有低吸水能力的高能共晶体。基于分子静电势的计算,发现了共晶体的共形成物中的分子间结合位点。通过晶格能量最小化方法,构造了可能的晶体结构,并预测了可能的高能共晶体的密度和氢键能。共晶筛选方法用于计算成对相互作用并选择有希望的共晶形成剂用于实验筛选。该方法可用于预测共晶体的吸湿性,从而可以加快新型高能共晶体材料的开发进度。基于分子静电势的计算,发现了共晶体的共形成物中的分子间结合位点。通过晶格能量最小化方法,构造了可能的晶体结构,并预测了可能的高能共晶体的密度和氢键能。共晶筛选方法用于计算成对相互作用并选择有希望的共晶形成剂用于实验筛选。该方法可用于预测共晶体的吸湿性,从而可以加快新型高能共晶体材料的开发进度。基于分子静电势的计算,发现了共晶体的共形成物中的分子间结合位点。通过晶格能量最小化方法,构造了可能的晶体结构,并预测了可能的高能共晶体的密度和氢键能。共晶筛选方法用于计算成对相互作用并选择有希望的共晶形成剂用于实验筛选。该方法可用于预测共晶体的吸湿性,从而可以加快新型高能共晶体材料的开发进度。并预测了可能的高能共晶体的密度和氢键能。共晶筛选方法用于计算成对相互作用并选择有希望的共晶形成剂用于实验筛选。该方法可用于预测共晶体的吸湿性,从而可以加快新型高能共晶体材料的开发进度。并预测了可能的高能共晶体的密度和氢键能。共晶筛选方法用于计算成对相互作用并选择有希望的共晶形成剂用于实验筛选。该方法可用于预测共晶体的吸湿性,从而可以加快新型高能共晶体材料的开发进度。
更新日期:2020-08-10
中文翻译:
调节吸湿性的高能二硝酰胺铵共晶体设计的分子模拟研究
共结晶技术是提高晶体性能的有效方法。二硝酰胺铵(ADN)是推进剂的重要成分。但是,ADN的高吸湿性限制了其应用。为了解决此问题,九种含能的共形成物,其中包含2,4,5,8,10,12-六硝基-2,4,6,8,10,12-六氮杂异纤锌矿型结构烷烃(CL-20),苯并三呋喃(BTF) ,1,3,5,7-四硝基-1,3,5,7-四唑烷(HMX),2,4,6-三硝基甲苯(TNT),丁烷-1,2,3,4-四硝酸四丁酯(ETN) ,六氢-1,3,5-三硝基-1,3,5-三嗪(RDX),1,3,5-三硝基苯(TNB),2,4,6-三硝基-N-甲基苯胺(MATNB)和1选择1,3,3-三硝基氮杂环丁烷(TNAZ)来预测共晶体的吸湿性,其中ADN与共形成物的摩尔比为1:1。提出了一种新颖的计算方法来有效地选择共形成物以设计具有低吸水能力的高能共晶体。基于分子静电势的计算,发现了共晶体的共形成物中的分子间结合位点。通过晶格能量最小化方法,构造了可能的晶体结构,并预测了可能的高能共晶体的密度和氢键能。共晶筛选方法用于计算成对相互作用并选择有希望的共晶形成剂用于实验筛选。该方法可用于预测共晶体的吸湿性,从而可以加快新型高能共晶体材料的开发进度。基于分子静电势的计算,发现了共晶体的共形成物中的分子间结合位点。通过晶格能量最小化方法,构造了可能的晶体结构,并预测了可能的高能共晶体的密度和氢键能。共晶筛选方法用于计算成对相互作用并选择有希望的共晶形成剂用于实验筛选。该方法可用于预测共晶体的吸湿性,从而可以加快新型高能共晶体材料的开发进度。基于分子静电势的计算,发现了共晶体的共形成物中的分子间结合位点。通过晶格能量最小化方法,构造了可能的晶体结构,并预测了可能的高能共晶体的密度和氢键能。共晶筛选方法用于计算成对相互作用并选择有希望的共晶形成剂用于实验筛选。该方法可用于预测共晶体的吸湿性,从而可以加快新型高能共晶体材料的开发进度。并预测了可能的高能共晶体的密度和氢键能。共晶筛选方法用于计算成对相互作用并选择有希望的共晶形成剂用于实验筛选。该方法可用于预测共晶体的吸湿性,从而可以加快新型高能共晶体材料的开发进度。并预测了可能的高能共晶体的密度和氢键能。共晶筛选方法用于计算成对相互作用并选择有希望的共晶形成剂用于实验筛选。该方法可用于预测共晶体的吸湿性,从而可以加快新型高能共晶体材料的开发进度。
































 京公网安备 11010802027423号
京公网安备 11010802027423号