当前位置:
X-MOL 学术
›
J. Mater. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Crystal structure, electronic structure and thermoelectric properties of β- and γ-Zn4Sb3 thermoelectrics: a (3 + 1)-dimensional superspace group approach
Journal of Materials Chemistry C ( IF 5.7 ) Pub Date : 2020-06-05 , DOI: 10.1039/d0tc01497d Shun Yoshioka 1, 2, 3, 4, 5 , Kei Hayashi 1, 2, 3, 4, 5 , Aisaku Yokoyama 1, 2, 3, 4, 5 , Wataru Saito 1, 2, 3, 4, 5 , Hezhang Li 1, 2, 3, 4, 5 , Tomohisa Takamatsu 1, 2, 3, 4, 5 , Yuzuru Miyazaki 1, 2, 3, 4, 5
Journal of Materials Chemistry C ( IF 5.7 ) Pub Date : 2020-06-05 , DOI: 10.1039/d0tc01497d Shun Yoshioka 1, 2, 3, 4, 5 , Kei Hayashi 1, 2, 3, 4, 5 , Aisaku Yokoyama 1, 2, 3, 4, 5 , Wataru Saito 1, 2, 3, 4, 5 , Hezhang Li 1, 2, 3, 4, 5 , Tomohisa Takamatsu 1, 2, 3, 4, 5 , Yuzuru Miyazaki 1, 2, 3, 4, 5
Affiliation
|
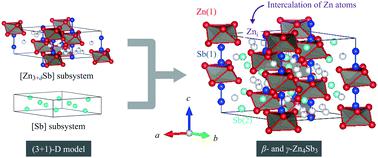
Thermoelectric (TE) materials are promising candidates for solving today's energy problem owing to their ability to directly convert waste heat into electricity via the Seebeck effect. One of the most efficient TE materials known is Zn4Sb3. To understand its high efficiency, a novel composite crystal structure model for β- and γ-phases of Zn4Sb3 is constructed using a (3 + 1)-dimensional ((3 + 1)-D) superspace group approach. This (3 + 1)-D model is expressed as [Zn3+δSb][Sb]p with the superspace group of R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m(00γ)0s. The [Zn3+δSb] and [Sb] subsystems have same a- and b-axis lengths but a different c-axis length. The (3 + 1)-D model contains four atomic sites: a Zn(1) normal site, an interstitial Zn (Zni) site and an Sb(1) site in the [Zn3+δSb] subsystem and an Sb(2) site in the [Sb] subsystem, which is different from a conventional 3D model containing additional Zni sites. The crystal structures of β- and γ-Zn4Sb3 are investigated via powder and synchrotron X-ray diffraction (XRD) measurements. The XRD patterns are well analysed by the (3 + 1)-D model. The occupancies of Zn(1), Sb(1) and Sb(2) sites are 100%, whereas the Zni occupancy changes depending on the heating time during the preparation of β-Zn4Sb3. Moreover, electronic density of states (DOS) of β-Zn4Sb3 with and without Zni is calculated based on the (3 + 1)-D model, demonstrating a close relationship between the DOS and the Zni occupancy. The calculated TE properties, such as Seebeck coefficient, electrical conductivity and power factor, also depend on the Zni occupancy.
m(00γ)0s. The [Zn3+δSb] and [Sb] subsystems have same a- and b-axis lengths but a different c-axis length. The (3 + 1)-D model contains four atomic sites: a Zn(1) normal site, an interstitial Zn (Zni) site and an Sb(1) site in the [Zn3+δSb] subsystem and an Sb(2) site in the [Sb] subsystem, which is different from a conventional 3D model containing additional Zni sites. The crystal structures of β- and γ-Zn4Sb3 are investigated via powder and synchrotron X-ray diffraction (XRD) measurements. The XRD patterns are well analysed by the (3 + 1)-D model. The occupancies of Zn(1), Sb(1) and Sb(2) sites are 100%, whereas the Zni occupancy changes depending on the heating time during the preparation of β-Zn4Sb3. Moreover, electronic density of states (DOS) of β-Zn4Sb3 with and without Zni is calculated based on the (3 + 1)-D model, demonstrating a close relationship between the DOS and the Zni occupancy. The calculated TE properties, such as Seebeck coefficient, electrical conductivity and power factor, also depend on the Zni occupancy.
中文翻译:

β-和γ-Zn4Sb3热电材料的晶体结构,电子结构和热电特性:(3 +1)维超空间群方法
由于热电(TE)材料能够通过塞贝克效应将废热直接转化为电能,因此有望成为解决当今能源问题的有力选择。已知的最有效的TE材料之一是Zn 4 Sb 3。为了了解其高效性,使用(3 +1)维((3 +1)-D)超空间群方法构造了Zn 4 Sb 3的β和γ相的新型复合晶体结构模型。此(3 + 1)-D模型被表示为[锌3+ δ锑] [锑] p与超空间组的ř米(00 γ)0小号。[Zn 3+ δ[Sb]和[Sb]子系统具有相同的a轴和b轴长度,但具有不同的c轴长度。在(3 + 1)-D模型包含四个原子位点:将Zn(1)正常部位,间隙的Zn(锌我)位点和在[Zn的锑(1)站点3+ δ锑]子系统和Sb的(2)[Sb]子系统中的站点,这与包含其他Zn i站点的常规3D模型不同。β-和γ-Zn系的晶体结构4的Sb 3进行了研究通过粉末和同步辐射X射线衍射(XRD)测量。通过(3 +1)-D模型可以很好地分析XRD模式。的Zn(1),锑(1)和Sb的占有率(2)位点是100%,而锌我占用β-Zn系的制备过程中根据加热时间而变化4的Sb 3。此外,β-Zn系的状态(DOS)的电子密度4的Sb 3具有和不具有锌我来计算基于(3 + 1)-D模型,展示出DOS和锌之间的密切关系我占用。计算出的TE特性(例如塞贝克系数,电导率和功率因数)也取决于Zn i的占有率。
更新日期:2020-07-16
m(00γ)0s. The [Zn3+δSb] and [Sb] subsystems have same a- and b-axis lengths but a different c-axis length. The (3 + 1)-D model contains four atomic sites: a Zn(1) normal site, an interstitial Zn (Zni) site and an Sb(1) site in the [Zn3+δSb] subsystem and an Sb(2) site in the [Sb] subsystem, which is different from a conventional 3D model containing additional Zni sites. The crystal structures of β- and γ-Zn4Sb3 are investigated via powder and synchrotron X-ray diffraction (XRD) measurements. The XRD patterns are well analysed by the (3 + 1)-D model. The occupancies of Zn(1), Sb(1) and Sb(2) sites are 100%, whereas the Zni occupancy changes depending on the heating time during the preparation of β-Zn4Sb3. Moreover, electronic density of states (DOS) of β-Zn4Sb3 with and without Zni is calculated based on the (3 + 1)-D model, demonstrating a close relationship between the DOS and the Zni occupancy. The calculated TE properties, such as Seebeck coefficient, electrical conductivity and power factor, also depend on the Zni occupancy.
中文翻译:
β-和γ-Zn4Sb3热电材料的晶体结构,电子结构和热电特性:(3 +1)维超空间群方法
由于热电(TE)材料能够通过塞贝克效应将废热直接转化为电能,因此有望成为解决当今能源问题的有力选择。已知的最有效的TE材料之一是Zn 4 Sb 3。为了了解其高效性,使用(3 +1)维((3 +1)-D)超空间群方法构造了Zn 4 Sb 3的β和γ相的新型复合晶体结构模型。此(3 + 1)-D模型被表示为[锌3+ δ锑] [锑] p与超空间组的ř
米(00 γ)0小号。[Zn 3+ δ[Sb]和[Sb]子系统具有相同的a轴和b轴长度,但具有不同的c轴长度。在(3 + 1)-D模型包含四个原子位点:将Zn(1)正常部位,间隙的Zn(锌我)位点和在[Zn的锑(1)站点3+ δ锑]子系统和Sb的(2)[Sb]子系统中的站点,这与包含其他Zn i站点的常规3D模型不同。β-和γ-Zn系的晶体结构4的Sb 3进行了研究通过粉末和同步辐射X射线衍射(XRD)测量。通过(3 +1)-D模型可以很好地分析XRD模式。的Zn(1),锑(1)和Sb的占有率(2)位点是100%,而锌我占用β-Zn系的制备过程中根据加热时间而变化4的Sb 3。此外,β-Zn系的状态(DOS)的电子密度4的Sb 3具有和不具有锌我来计算基于(3 + 1)-D模型,展示出DOS和锌之间的密切关系我占用。计算出的TE特性(例如塞贝克系数,电导率和功率因数)也取决于Zn i的占有率。
































 京公网安备 11010802027423号
京公网安备 11010802027423号