当前位置:
X-MOL 学术
›
Mol. Pharmaceutics
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
药物熔解特性的机器估计及其对溶解度预测的影响。
Molecular Pharmaceutics ( IF 4.5 ) Pub Date : 2020-06-04 , DOI: 10.1021/acs.molpharmaceut.0c00355
Nicole Wyttenbach 1 , Andreas Niederquell 2 , Martin Kuentz 2
Molecular Pharmaceutics ( IF 4.5 ) Pub Date : 2020-06-04 , DOI: 10.1021/acs.molpharmaceut.0c00355
Nicole Wyttenbach 1 , Andreas Niederquell 2 , Martin Kuentz 2
Affiliation
|
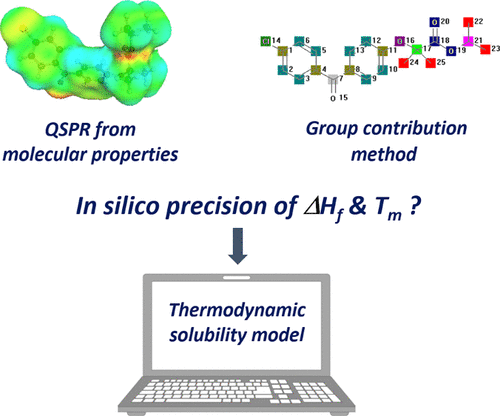
最近人们对机器学习(ML)和分子定量结构特性关系(QSPR)产生了浓厚的兴趣。本研究评估了在商业软件(COSMOquick和Molecular Modeling Pro)中实现的基于ML的现代方法,与传统的基团贡献方法(Joback和Reid方法)相比,可估计熔点和熔融焓。从文献中收集了广泛的市场化合物数据,以及通过差示扫描量热法对候选药物进行了测量的新数据。使用随机梯度提升的QSPR可以实现最高的预测精度。讨论了模型偏差,尤其是对热力学溶解度建模的影响,因为这通常需要估计熔点和熔融焓。结果表明,尽管预测准确性有了很大的提高,但仍然存在局限性,尤其是对于复杂的候选药物。建议在这种情况下,获得熔融性能应谨慎使用in silico作为热力学溶解度建模的输入数据。未来的研究将显示如何通过更大的数据集和其他ML算法或通过使用分子模拟来进一步提高热物理药物特性的预测极限。

"点击查看英文标题和摘要"
更新日期:2020-07-06
"点击查看英文标题和摘要"
































 京公网安备 11010802027423号
京公网安备 11010802027423号